
Efficacy and Safety of Eculizumab in Enteroaggregative E. coli Associated Hemolytic Uremic Syndrome


Abstract
1. Introduction
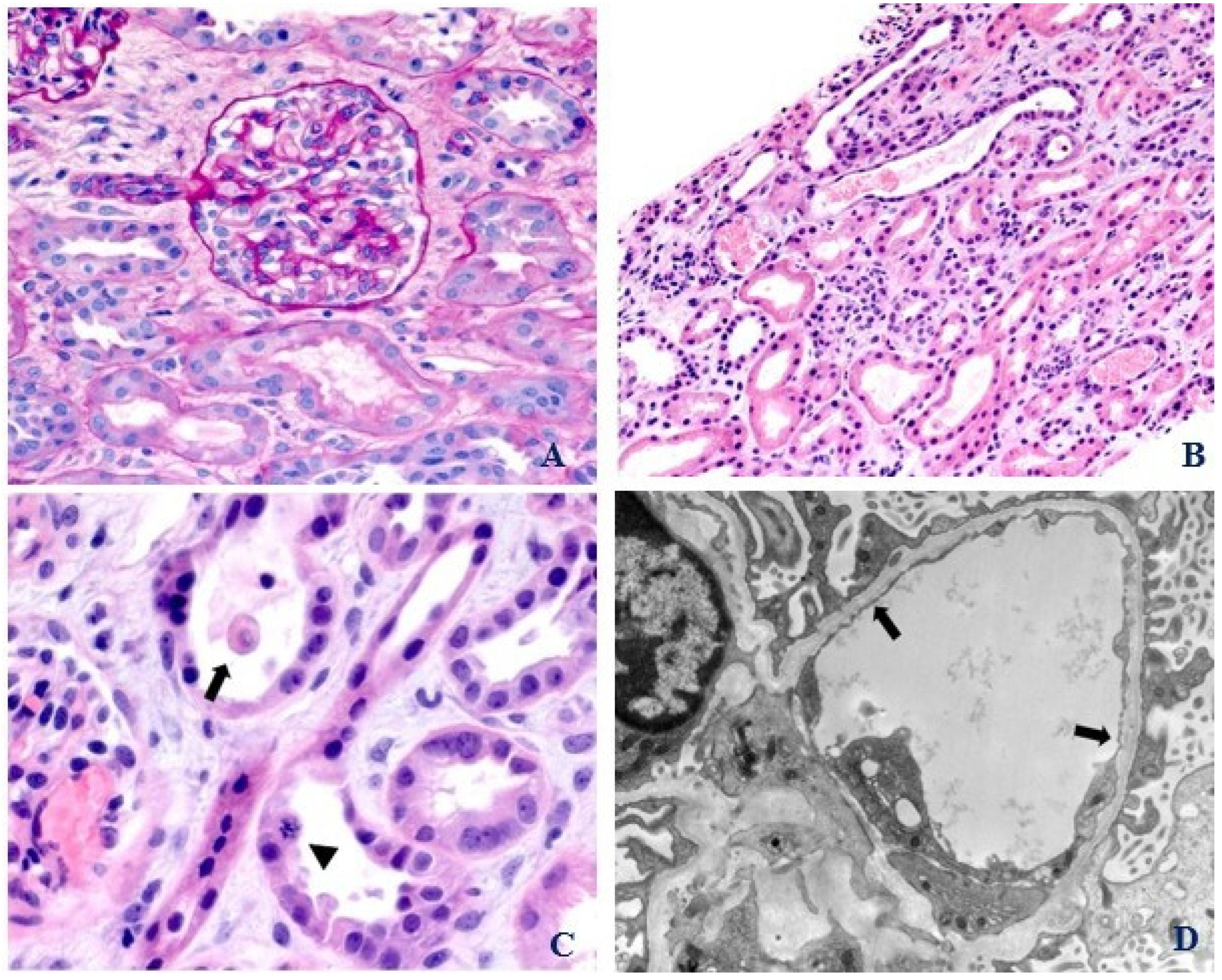
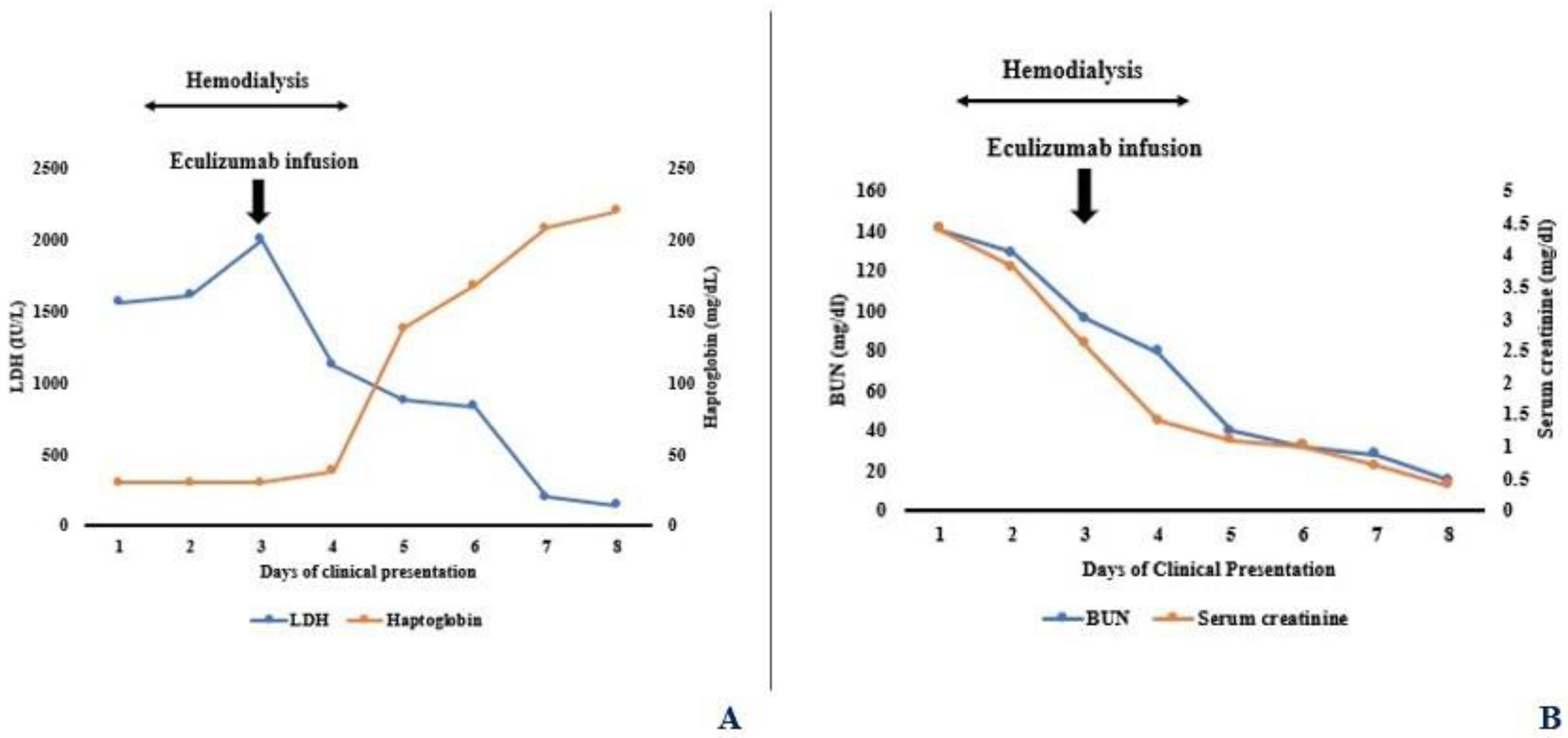
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Noris, M.; Remuzzi, G. Atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Sallée, M.; Ismail, K.; Fakhouri, F.; Vacher-Coponat, H.; Moussi-Francés, J.; Frémaux-Bacchi, V.; Burtey, S. Thrombocytopenia is not mandatory to diagnose haemolytic and uremic syndrome. BMC Nephrol. 2013, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Kielstein, J.T.; Beutel, G.; Fleig, S.; Steinhoff, J.; Meyer, T.N.; Hafer, C.; Kuhlmann, U.; Bramstedt, J.; Panzer, U.; Vischedyk, M.; et al. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: An analysis of the German STEC-HUS registry. Nephrol. Dial. Transplant. 2012, 27, 3807–3815. [Google Scholar] [CrossRef] [PubMed]
- Law, D.; Chart, H. Enteroaggregative Escherichia coli. J. Appl. Microbiol. 1998, 84, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Okeke, I.N.; Nataro, J.P. Enteroaggregative Escherichia coli. Lancet Infect. Dis. 2001, 1, 304–313. [Google Scholar] [CrossRef]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef]
- Lapeyraque, A.L.; Malina, M.; Fremeaux-Bacchi, V.; Boppel, T.; Kirschfink, M.; Oualha, M.; Proulx, F.; Clermont, M.-J.; Le Deist, F.; Niaudet, P.; et al. Eculizumab in severe Shiga-toxin-associated HUS. N. Engl. J. Med. 2011, 364, 2561–2563. [Google Scholar] [CrossRef] [PubMed]
- Giordano, P.; Netti, G.S.; Santangelo, L.; Castellano, G.; Carbone, V.; Torres, D.D.; Martino, M.; Sesta, M.; Di Cuonzo, F.; Resta, M.C.; et al. A pediatric neurologic assessment score may drive the eculizumab-based treatment of Escherichia coli-related hemolytic uremic syndrome with neurological involvement. Pediatr. Nephrol. 2019, 34, 517–527. [Google Scholar] [CrossRef]
- Diurno, F.; Numis, F.G.; Porta, G.; Cirillo, F.; Maddaluno, S.; Ragozzino, A.; De Negri, P.; Di Gennaro, C.; Pagano, A.; Allegorico, E.; et al. Eculizumab treatment in patients with COVID-19: Preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4040–4047. [Google Scholar]
- Matošević, M.; Kos, I.; Davidović, M.; Ban, M.; Matković, H.; Jakopčić, I.; Vuković Brinar, I.; Szilágyi, Á.; Csuka, D.; Sinkovits, G.; et al. Hemolytic uremic syndrome in the setting of COVID-19 successfully treated with complement inhibition therapy: An instructive case report of a previously healthy toddler and review of literature. Front. Pediatr. 2023, 11, 1092860. [Google Scholar] [CrossRef]
- Tanaka, K.; Adams, B.; Aris, A.M.; Fujita, N.; Ogawa, M.; Ortiz, S.; Vallee, M.; Greenbaum, L.A. The long-acting C5 inhibitor, ravulizumab, is efficacious and safe in pediatric patients with atypical hemolytic uremic syndrome previously treated with eculizumab. Pediatr. Nephrol. 2021, 36, 889–898. [Google Scholar] [CrossRef]
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar] [CrossRef]
- De Serres, S.A.; Isenring, P. Athrombocytopenic thrombotic microangiopathy, a condition that could be overlooked based on current diagnostic criteria. Nephrol. Dial. Transplant. 2009, 24, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, G.; Leung, N.; Said, S.M.; Valeri, A.M.; Astor, B.C.; Fidler, M.E.; Alexander, M.P.; Cornell, L.D.; Nasr, S.H. The prevalence and clinical outcomes of microangiopathic hemolytic anemia in patients with biopsy-proven renal thrombotic microangiopathy. Am. J. Hematol. 2022, 97, E426–E429. [Google Scholar] [CrossRef] [PubMed]
- Balestracci, A.; Toledo, I.; Meni Battaglia, L.; de Lillo, L.; More, N.; Cao, G.; Alvarado, C. Postdiarrhoeal haemolytic uraemic syndrome without thrombocytopenia. Nefrologia 2017, 37, 508–514. [Google Scholar] [CrossRef]
- Sellier-Leclerc, A.L.; Fremeaux-Bacchi, V.; Dragon-Durey, M.A.; Macher, M.-A.; Niaudet, P.; Guest, G.; Boudailliez, B.; Bouissou, F.C.; Deschenes, G.; Gie, S.; et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2007, 18, 2392–2400. [Google Scholar] [CrossRef]
- Murphree, C.R.; Nguyen, N.N.; Shatzel, J.J.; Olson, S.R.; Chung, P.D.; Lockridge, J.B.; Andeen, N.K.; DeLoughery, T.G. Biopsy-proven thrombotic microangiopathy without schistocytosis on peripheral blood smear: A cautionary tale. Am. J. Hematol. 2019, 94, E234–E237. [Google Scholar] [CrossRef] [PubMed]
- Genest, D.S.; Patriquin, C.J.; Licht, C.; John, R.; Reich, H.N. Renal Thrombotic Microangiopathy: A Review. Am. J. Kidney Dis. 2023, 81, 591–605. [Google Scholar] [CrossRef]
- Estrada, C.C.; Maldonado, A.; Mallipattu, S.K. Therapeutic Inhibition of VEGF Signaling and Associated Nephrotoxicities. J. Am. Soc. Nephrol. 2019, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, D.; Goodship, T.H.; Richards, A. Atypical hemolytic uremic syndrome. Semin. Nephrol. 2013, 33, 508–530. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C. Membranoproliferative glomerulonephritis—A new look at an old entity. N. Engl. J. Med. 2012, 366, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Goodship, T.H.; Cook, H.T.; Fakhouri, F.; Fervenza, F.C.; Frémeaux-Bacchi, V.; Kavanagh, D.; Nester, C.M.; Noris, M.; Pickering, M.C.; Rodríguez de Córdoba, S.; et al. Conference Participants. Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M.; Marians, R.; Emlen, W.; Wood, S.; Smith, C.; Akana, H.; Holers, V.M.; Lesser, M.; Kline, M.; Hoffman, C.; et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin. J. Am. Soc. Nephrol. 2009, 4, 1920–1924. [Google Scholar] [CrossRef] [PubMed]
- Poolpol, K.; Orth-Höller, D.; Speth, C.; Zipfel, P.F.; Skerka, C.; de Córdoba, S.R.; Brockmeyer, J.; Bielaszewska, M.; Würzner, R. Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol. Immunol. 2014, 58, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Delmas, Y.; Vendrely, B.; Clouzeau, B.; Bachir, H.; Bui, H.-N.; Lacraz, A.; Hélou, S.; Bordes, C.; Reffet, A.; Llanas, B.; et al. Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: Outcome with eculizumab. Nephrol. Dial. Transplant. 2014, 29, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Abreu, A.G.; Barbosa, A.S. How Escherichia coli circumvent complement-mediated killing. Front. Immunol. 2017, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Hill, A.; Hill, Q.A.; Tvedt, T.H.A.; Michel, M. Novel insights into the treatment of complement-mediated hemolytic anemias. Ther. Adv. Hematol. 2019, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Puri, V.; Gandhi, A.; Sharma, S. Renal biopsy in paroxysmal nocturnal hemoglobinuria: An insight into the spectrum of morphologic changes. Indian J. Nephrol. 2017, 27, 284–288. [Google Scholar]
- Varela, J.C.; Tomlinson, S. Complement: An overview for the clinician. Hematol. Oncol. Clin. N. Am. 2015, 29, 409–427. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Investigations | Values | Normal Values |
|---|---|---|
| WBC count | 17.3 × 109/L | 4–11 × 109/L |
| Hemoglobin | 6.3 g/dL | 10.5–13.5 g/dL |
| Platelet count | 343 × 109/L | 150–450 × 109/L |
| LDH | 1565 IU/L | 135–225 IU/L |
| Haptoglobin | <30 mg/dL | 40–215 mg/dL |
| Peripheral smear | Absence of schistocytes, spherocytes | |
| Direct antiglobulin test, C3 and IgG | Negative | |
| ADAMTS13 activity | 80% | >60% |
| C3 complement | 95 mg/dL | 87–200 mg/dL |
| C4 complement | 21 mg/dL | 13–50 mg/dL |
| SC5B-9 level | 469 ng/mL | ≤244 ng/mL |
| Genetic testing for atypical HUS | Negative for complement mutations or deficiencies | |
| Uric acid | 12 mg/dL | 2.6–6.8 mg/dL |
| Serum sodium | 134 mmol/L | 135–145 mmol/L |
| Serum potassium | 5.4 mmol/L | 3.5–4.5 mmol/L |
| Serum bicarbonate | 12 mmol/L | 22–30 mmol/L |
| BUN | 114 mg/dL | 6–21 mg/dL |
| Serum creatinine | 3.1 mg/dL | 0.20–0.43 mg/dL |
| Serum calcium | 8.7 mg/dL | 8.4–10.2 mg/dL |
| Serum phosphorus | 7/8 mg/dL | 4.3–6.8 mg/dL |
| Serum albumin | 3.2 g/dL | 3.5–5.2 g/dL |
| ALT, AST | 23 IU/L, 118 IU/L | 0–35 IU/L, 0–37 IU/L |
| Total serum bilirubin | 1.8 mg/dL | 0–1 mg/dL |
| Sickle cell screen | Negative | |
| Serum folate and vitamin B12 levels | Normal | |
| G6PD deficiency | Absent | |
| Respiratory virus panel, including SARS-CoV-2 PCR | Negative | |
| EBV DNA PCR, CMV DNA PCR | Negative | |
| PTH | 105 pg/mL | 12–88 pg/mL |
| Iron saturation | 41% | 20–55% |
| Stool test | Positive for EAEC, negative for Shiga-toxin | |
| Urinalysis | 2 RBC/hpf, 2 WBC/hpf, trace proteinuria, negative nitrites and leukocytes, pH 7, specific gravity 1.025 | |
| Plasma free hemoglobin | 190 mg/dL | <150 mg/dL |
| CRP | 44 mg/L | 0–5 mg/L |
| Procalcitonin | 36.37 ng/mL | <0.1 ng/mL |
| Cultures, blood, and urine | Negative | |
| CK | 161 U/L | 0–180 U/L |
| ANA, ANCA, Anti-GBM antibody | Negative | |
| Renal bladder sonogram | Enlarged, echogenic kidneys measuring 9–9.5 cm in length, normal Doppler examination with no evidence of renal vein thrombosis | |
| Abdominal sonogram | No splenomegaly or hepatomegaly |
| Classic HUS | Partial HUS | Index Case | |
|---|---|---|---|
| Anemia | Present | Present | Present |
| Thrombocytopenia | Present | May be absent | Absent |
| Peripheral schistocytes | Present | May be absent | Absent |
| LDH | Elevated | Elevated | Elevated |
| Haptoglobin | Low | Low | Low/Undetected |
| Acute Kidney Injury | Present | Present | Present |
| Renal pathologic lesions of classic TMA | Present | May be absent | Absent |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acharya, R.; Clapp, W.L.; Upadhyay, K. Efficacy and Safety of Eculizumab in Enteroaggregative E. coli Associated Hemolytic Uremic Syndrome. Pediatr. Rep. 2024, 16, 26-34. https://doi.org/10.3390/pediatric16010003
Acharya R, Clapp WL, Upadhyay K. Efficacy and Safety of Eculizumab in Enteroaggregative E. coli Associated Hemolytic Uremic Syndrome. Pediatric Reports. 2024; 16(1):26-34. https://doi.org/10.3390/pediatric16010003
Chicago/Turabian StyleAcharya, Ratna, William L. Clapp, and Kiran Upadhyay. 2024. "Efficacy and Safety of Eculizumab in Enteroaggregative E. coli Associated Hemolytic Uremic Syndrome" Pediatric Reports 16, no. 1: 26-34. https://doi.org/10.3390/pediatric16010003
APA StyleAcharya, R., Clapp, W. L., & Upadhyay, K. (2024). Efficacy and Safety of Eculizumab in Enteroaggregative E. coli Associated Hemolytic Uremic Syndrome. Pediatric Reports, 16(1), 26-34. https://doi.org/10.3390/pediatric16010003