Pediatric Autoimmune Hepatitis


Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maggiore, G.; Nastasio, S.; Sciveres, M. Juvenile autoimmune hepatitis: Spectrum of the disease. World J. Hepatol. 2014, 6, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Mieli-Vergani, G.; Vergani, D.; Baumann, U.; Czubkowski, P.; Debray, D.; Dezsofi, A.; Fischler, B.; Gupte, G.; Hierro, L.; Indolfi, G.; et al. Diagnosis and Management of Pediatric Autoimmune Liver Disease: ESPGHAN Hepatology Committee Position Statement. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.L.; Adams, D.; Assis, D.N.; Kerkar, N.; Manns, M.P.; Mayo, M.J.; Vierling, J.M.; Alsawas, M.; Murad, M.H.; Czaja, A.J. Diagnosis and Management of Autoimmune Hepatitis in Adults and Children: 2019 Practice Guidance and Guidelines from the American Association for the Study of Liver Diseases. Hepatology 2020, 72, 671–722. [Google Scholar] [CrossRef] [PubMed]
- Sciveres, M.; Nastasio, S.; Maggiore, G. Novel Diagnostic and Therapeutic Strategies in Juvenile Autoimmune Hepatitis. Front. Pediatr. 2019, 7, 382. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.M.; Zeniya, M.; Czaja, A.J.; Parés, A.; Dalekos, G.N.; Krawitt, E.L.; Bittencourt, P.L.; Porta, G.; Boberg, K.M.; Hofer, H.; et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008, 48, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.; Berg, P.A.; Bianchi, F.B.; Bianchi, L.; Burroughs, A.K.; Cancado, E.L.; Chapman, R.W.; Cooksley, W.G.E.; Czaja, A.J.; Desmet, V.J.; et al. International Autoimmune Hepatitis Group Report: Review of criteria for diagnosis of autoimmune hepatitis. J. Hepatol. 1999, 31, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Arcos-Machancoses, J.V.; Busoms, C.M.; Tatis, E.J.; Bovo, M.V.; de Carpi, J.M. Accuracy of the Simplified Criteria for Autoimmune Hepatitis in Children: Systematic Review and Decision Analysis. J. Clin. Exp. Hepatol. 2019, 9, 147–155. [Google Scholar] [CrossRef] [PubMed]
- van Gerven, N.M.; Verwer, B.J.; Witte, B.I.; van Hoek, B.; Coenraad, M.J.; van Erpecum, K.J.; Beuers, U.; van Buuren, H.R.; de Man, R.A.; Drenth, J.P.; et al. Relapse is almost universal after withdrawal of immunosuppressive medication in patients with autoimmune hepatitis in remission. J. Hepatol. 2013, 58, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Deneau, M.; Book, L.S.; Guthery, S.L.; Jensen, M.K. Outcome after Discontinuation of Immunosuppression in Children with Autoimmune Hepatitis: A Population-Based Study. J. Pediatr. 2014, 164, 714–719.e2. [Google Scholar] [CrossRef] [PubMed]
- Saadah, O.I.; Smith, A.L.; Hardikar, W. Long-term outcome of autoimmune hepatitis in children. J. Gastroenterol. Hepatol. 2001, 16, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, G.V.; Portmann, B.; Reid, F.; Donaldson, P.T.; Doherty, D.G.; McCartney, M.; Mowat, A.P.; Vergani, D.; Mieli-Vergani, G. Autoimmune hepatitis in childhood: A 20-year experience. Hepatology 1997, 25, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Maggiore, G.; Bernard, O.; Mosca, A.; Ballot, E.; Johanet, C.; Jacquemin, E. Long-term outcomes of patients with type 1 or 2 autoimmune hepatitis presenting in childhood. J. Hepatol. 2023, 78, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Lohse, A.W.; Hørby Jørgensen, M.J. Paediatric autoimmune hepatitis: Time to change the textbooks? J. Hepatol. 2023, 78, 893–895. [Google Scholar] [CrossRef] [PubMed]
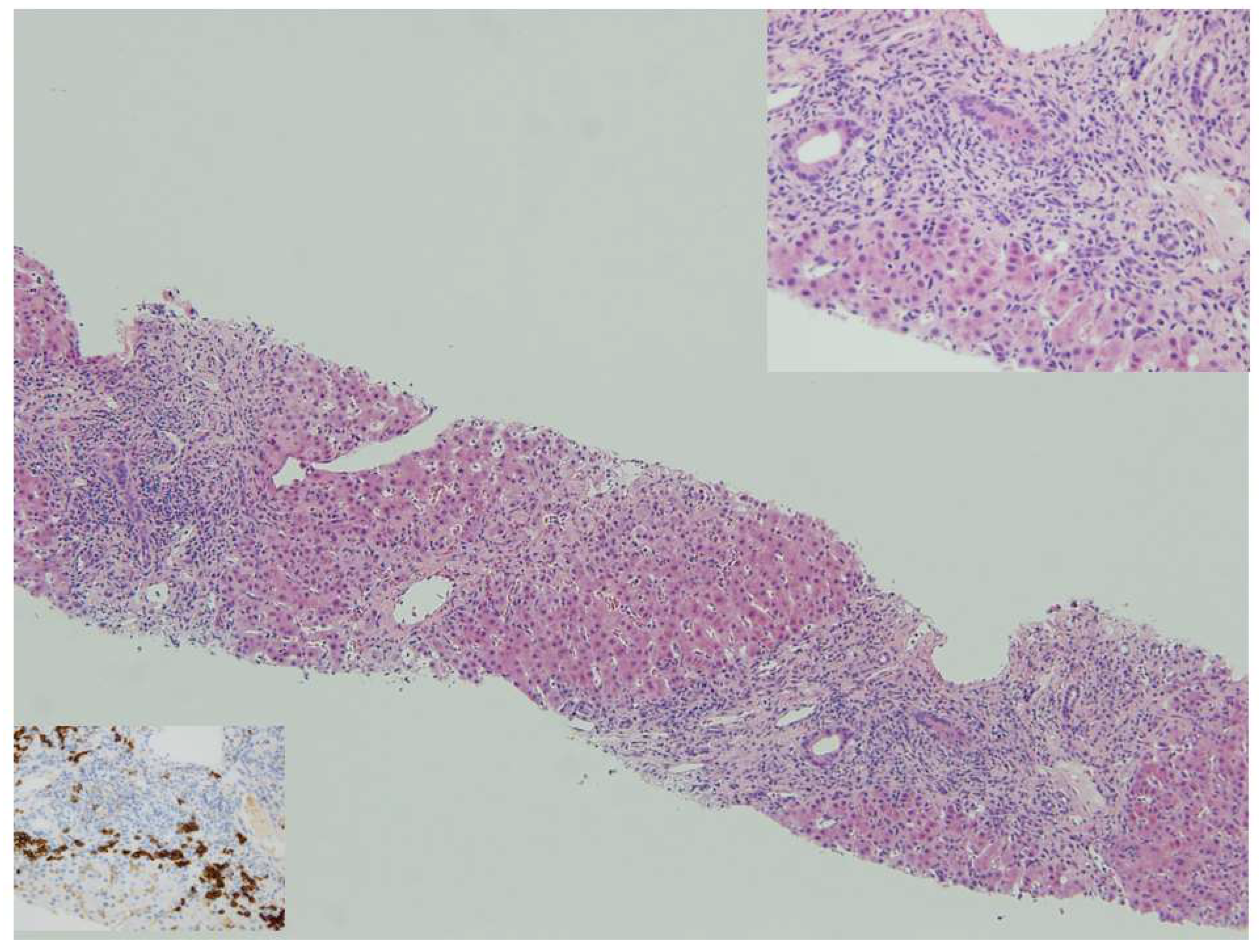

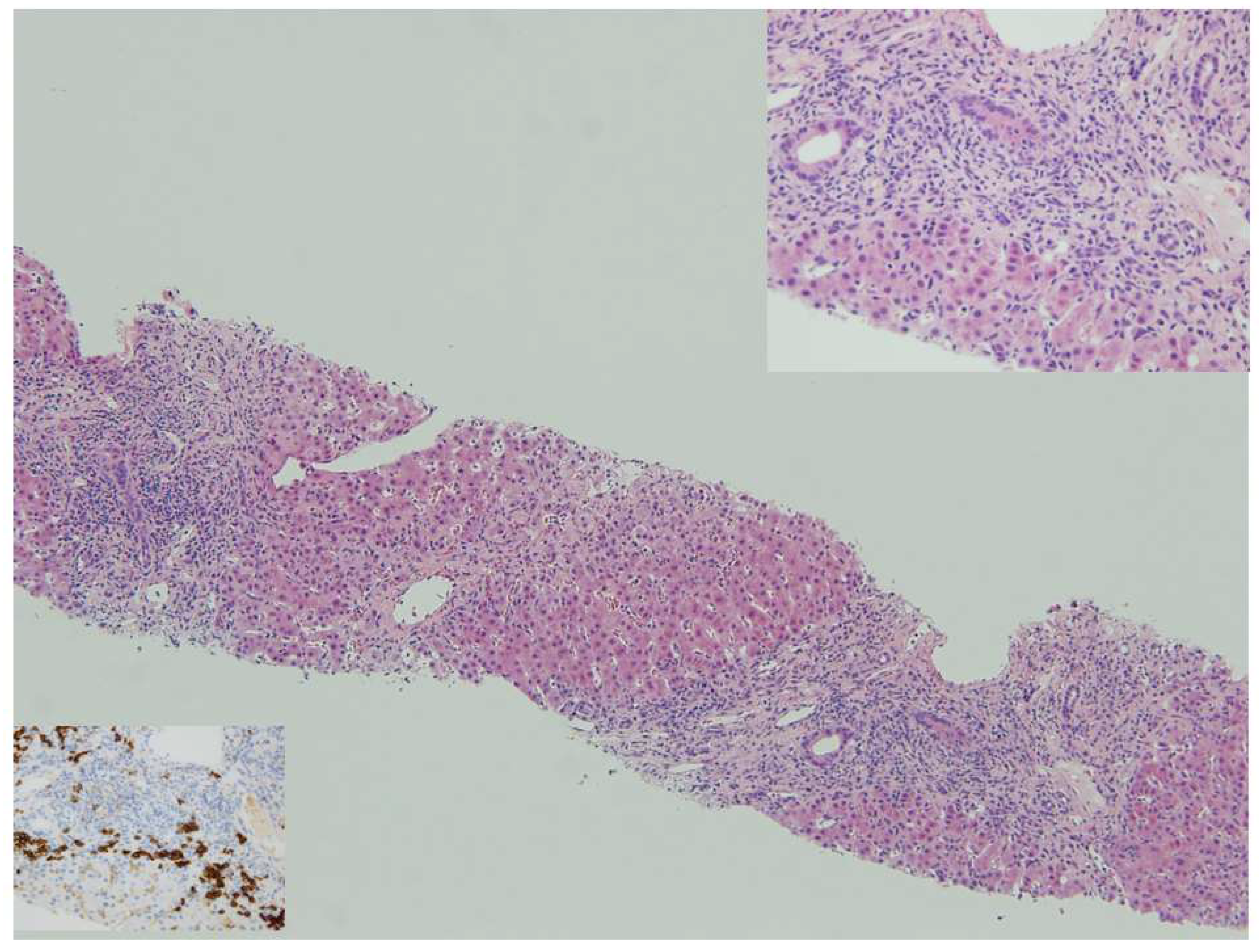{kind=link}
| Type of Presentation | Clinical Characteristics | Frequency | Additional Notes |
|---|---|---|---|
| Acute onset | Anorexia, nausea, vomiting, and abdominal pain followed by jaundice, similar to acute viral hepatitis. | Most common presentation | Some patients go on to develop acute liver failure with encephalopathy |
| Insidious onset | Progressive fatigue, anorexia, and intermittent jaundice lasting for several months/years before diagnosis | ~30% of patients | All patients have clinical evidence of chronic liver disease and/or cirrhosis at diagnosis |
| Asymptomatic | No symptoms | ~10–15% of patients | Incidental finding of clinical signs of chronic liver disease or elevated transaminases |
| Symptomatic portal hypertension | Variceal bleed; ascites | Rare | |
| JIAH presenting with symptoms related to an extrahepatic autoimmune disease | Symptoms related to autoimmune thrombocytopenia, autoimmune hemolytic anemia, type 1 diabetes, autoimmune thyroiditis, vitiligo, cutaneous vasculitis, uveitis, glomerulonephritis, juvenile chronic arthritis, systemic lupus erythematosus, Sjögren’s syndrome, celiac disease, and inflammatory bowel disease | Rare |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nastasio, S.; Sciveres, M.; Francalanci, P.; Maggiore, G. Pediatric Autoimmune Hepatitis. Pediatr. Rep. 2024, 16, 110-113. https://doi.org/10.3390/pediatric16010011
Nastasio S, Sciveres M, Francalanci P, Maggiore G. Pediatric Autoimmune Hepatitis. Pediatric Reports. 2024; 16(1):110-113. https://doi.org/10.3390/pediatric16010011
Chicago/Turabian StyleNastasio, Silvia, Marco Sciveres, Paola Francalanci, and Giuseppe Maggiore. 2024. "Pediatric Autoimmune Hepatitis" Pediatric Reports 16, no. 1: 110-113. https://doi.org/10.3390/pediatric16010011
APA StyleNastasio, S., Sciveres, M., Francalanci, P., & Maggiore, G. (2024). Pediatric Autoimmune Hepatitis. Pediatric Reports, 16(1), 110-113. https://doi.org/10.3390/pediatric16010011