
Gut Microbiome Structural Dynamics in Japanese Quail Across Developmental Stages

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
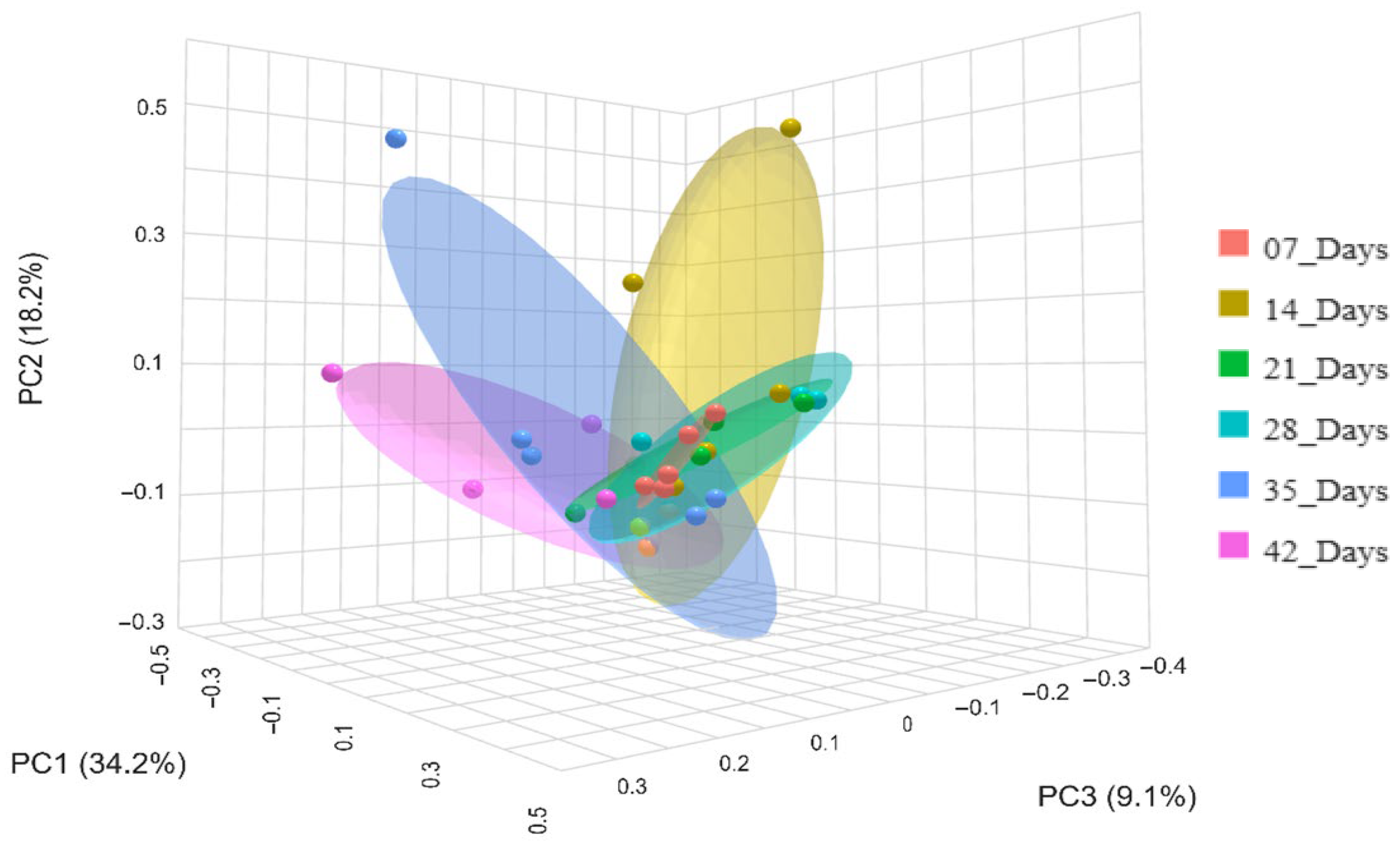
3. Results
3.1. Relative Abundance
3.2. Microbiota Diversity
3.3. Differential Abundance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silva, A.F.; Sgavioli, S.; Domingues, C.H.F.; Garcia, R.G. Quail farming as an alternative to increase the income of small producers. Arq. Bras. Med. Vet. Zootec. 2018, 70, 913–920. [Google Scholar] [CrossRef]
- Rinttilä, T.; Apajalathi, J. Intestinal microbiota and metabolites—Implications for broiler chicken health and performance. World’s Poult. Sci. J. 2013, 22, 647–658. [Google Scholar] [CrossRef]
- Ocejo, M.; Oporto, B.; Hurtado, A. 16S rRNA amplicon sequencing characterization of caecal microbiome composition of broilers and free-range slow-growing chickens throughout their productive lifespan. Sci. Rep. 2019, 9, 2506. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Chen, J.; Liu, K.; Tang, M.; Yang, Y. The avian gut microbiota: Diversity, influencing factors, and future directions. Front. Microbiol. 2022, 13, 934272. [Google Scholar] [CrossRef]
- Díaz Carrasco, J.M.; Casanova, N.A.; Fernández Miyakawa, M.E. Microbiota, gut health and chicken productivity: What is the connection? Microorganisms 2019, 7, 374. [Google Scholar] [CrossRef]
- Pan, D.; Yu, Z. Intestinal microbiome of poultry and its interaction with host and diet. Gut Microbes 2013, 5, 108–119. [Google Scholar] [CrossRef]
- Waite, D.W.; Taylor, M.W. Characterizing the avian gut microbiota: Membership, driving influences, and potential function. Front. Microbiol. 2014, 5, 223. [Google Scholar] [CrossRef]
- Choi, K.Y.; Lee, T.K.; Sul, W.J. Metagenomic analysis of chicken gut microbiota for improving metabolism and health of chickens—A review. Asian-Australas. J. Anim. Sci. 2015, 28, 1217–1225. [Google Scholar] [CrossRef]
- Wei, S.; Morrison, M.; Yu, Z. Bacterial census of poultry intestinal microbiome. Poult. Sci. 2013, 92, 671–683. [Google Scholar] [CrossRef]
- Wilkinson, T.J.; Cowan, A.A.; Vallin, H.E.; Onime, L.A.; Oyama, L.B.; Cameron, S.J.; Gonot, C.; Moorby, J.M.; Waddams, K.; Theobald, V.J.; et al. Characterization of the microbiome along the gastrointestinal tract of growing turkeys. Front. Microbiol. 2017, 8, 1089. [Google Scholar] [CrossRef]
- Gong, J.; Forster, R.J.; Yu, H.; Chambers, J.R.; Sabour, P.M.; Wheatcroft, R.; Chen, S. Diversity and phylogenetic analysis of bacteria in the mucosa of chicken ceca and comparison with bacteria in the cecal lumen. FEMS Microbiol. Lett. 2002, 208, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Brulc, J.M.; Wilson, M.K.; Law, B.F.; Theoret, J.R.; Joens, L.A.; Konkel, M.E.; Angly, F.; Dinsdale, E.A.; Edwards, R.A.; et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ONE 2008, 3, e2945. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Hughes, R.J.; Aspden, W.J.; Chapman, J.; Moore, R.J.; Stanley, D. The gastrointestinal tract microbiota of the Japanese quail, Coturnix japonica. Appl. Microbiol. Biotechnol. 2016, 100, 4201–4209. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.E.; Xiong, X.W.; Xu, J.G.; Gong, J.S.; Li, J.; Xu, Q.; Li, Y.F.; Yang, Y.B.; Zhou, M.; Zhu, X.N.; et al. Metagenomic analysis identifies sex-related cecal microbial gene functions and bacterial taxa in the quail. Front. Vet. Sci. 2021, 8, 693755. [Google Scholar] [CrossRef]
- Liu, S.; Tun, H.M.; Leung, F.C.; Bennett, D.C.; Zhang, H.; Cheng, K.M. Interaction of genotype and diet on small intestine microbiota of Japanese quail fed a cholesterol enriched diet. Sci. Rep. 2018, 8, 2381. [Google Scholar] [CrossRef]
- Du, X.; Xiang, Y.; Lou, F.; Tu, P.; Zhang, X.; Hu, X.; Lyu, W.; Xiao, Y. Microbial community and short-chain fatty acid mapping in the intestinal tract of quail. Animals 2020, 10, 1006. [Google Scholar] [CrossRef]
- Wilkinson, N.; Hughes, R.J.; Bajagai, Y.S.; Aspden, W.J.; Hao Van, T.T.; Moore, R.J.; Stanley, D. Reduced environmental bacterial load during early development and gut colonisation has detrimental health consequences in Japanese quail. Heliyon 2020, 6, e03213. [Google Scholar] [CrossRef]
- Silva, J.H.V.; Costa, F.G.P. Tabela para Codornas Japonesas e Europeias, 2nd ed.; FUNEP: Jaboticabal, Brazil, 2009. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, G.; Ewald, J.; Pang, Z.; Shiri, T.; Xia, J. MicrobiomeAnalyst 2.0: Comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res. 2023, 51, W310–W318. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Joly, C.; Gay-Quéheillard, J.; Léké, A.; Chardon, K.; Delanaud, S.; Bach, V.; Khorsi-Cauet, H. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME®) and in the rat. Environ. Sci. Pollut. Res. 2013, 20, 2726–2734. [Google Scholar] [CrossRef] [PubMed]
- Crisol-Martínez, E.; Moreno-Moyano, L.T.; Wilkinson, N.; Prasai, T.; Brown, P.H.; Moore, R.J.; Stanley, D. A low dose of an organophosphate insecticide causes dysbiosis and sex-dependent responses in the intestinal microbiota of the Japanese quail (Coturnix japonica). PeerJ 2016, 4, e2002. [Google Scholar] [CrossRef]
- de Vos, W.M.; de Vos, E.A.J. Role of the intestinal microbiome in health and disease: From correlation to causation. Nutr. Rev. 2012, 70 (Suppl. 1), S45–S56. [Google Scholar] [CrossRef]
- Alexander, M.; Ang, Q.Y.; Nayak, R.R.; Bustion, A.E.; Sandy, M.; Zhang, B.; Upadhyay, V.; Pollard, K.S.; Lynch, S.V.; Turnbaugh, P.J. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe 2022, 30, 17–30 e9. [Google Scholar] [CrossRef] [PubMed]
- Kraimi, N.; Calandreau, L.; Biesse, M.; Rabot, S.; Guitton, E.; Velge, P.; Leterrier, C. Absence of gut microbiota reduces emotional reactivity in Japanese quails (Coturnix japonica). Front. Physiol. 2018, 9, 603. [Google Scholar] [CrossRef]
- Rossi, M.; Amaretti, A.; Raimondi, S. Folate production by probiotic bacteria. Nutrients 2011, 3, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Oakley, B.B.; Lillehoj, H.S.; Kogut, M.H.; Kim, W.K.; Maurer, J.J.; Pedroso, A.; Lee, M.D.; Collett, S.R.; Johnson, T.J.; Cox, N.A. The chicken gastrointestinal microbiome. FEMS Microbiol. Lett. 2014, 360, 100–112. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Woting, A.; Clavel, T.; Loh, G.; Blaut, M. Bacterial transformation of dietary lignans in gnotobiotic rats. FEMS Microbiol. Ecol. 2010, 72, 507–514. [Google Scholar] [CrossRef]
- Li, S.; Lin, R.; Chen, J.; Hussain, R.; Zhang, S.; Su, Y.; Chan, Y.; Ghaffar, A.; Shi, D. Integrated gut microbiota and metabolomic analysis reveals immunomodulatory effects of Echinacea extract and Astragalus polysaccharides. Front. Vet. Sci. 2022, 9, 971058. [Google Scholar] [CrossRef]
- Shang, Y.; Kumar, S.; Oakley, B.; Kim, W.K. Chicken gut microbiota: Importance and detection technology. Front. Vet. Sci. 2018, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Stanley, D.; Denman, S.E.; Hughes, R.J.; Geier, M.S.; Crowley, T.M.; Chen, H.; Haring, V.R.; Moore, R.J. Intestinal microbiota associated with differential feed conversion efficiency in chickens. Appl. Microbiol. Biotechnol. 2012, 96, 1361–1369. [Google Scholar] [CrossRef]
- Tailford, L.E.; Crost, E.H.; Kavanaugh, D.; Juge, N. Mucin glycan foraging in the human gut microbiome. Front. Genet. 2015, 6, 81. [Google Scholar] [CrossRef]
- Yang, J.; Qin, K.; Sun, Y.; Yang, X. Microbiota-accessible fiber activates short-chain fatty acid and bile acid metabolism to improve intestinal mucus barrier in broiler chickens. Microbiol. Spectr. 2024, 12, e0206523. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, J.; Guo, X.; Xu, H.; Huang, C.; Nie, Y.; Zhou, Y. Odoribacter splanchnicus—A next-generation probiotic candidate. Microorganisms 2025, 13, 815. [Google Scholar] [CrossRef] [PubMed]
- Bajagai, Y.S.; Van, T.T.H.; Joat, N.; Chousalkar, K.; Moore, R.J.; Stanley, D. Layer chicken microbiota: A comprehensive analysis of spatial and temporal dynamics across all major gut sections. J. Anim. Sci. Biotechnol. 2024, 15, 20. [Google Scholar] [CrossRef]
- Inan, M.S.; Tolmacheva, V.; Wang, Q.S.; Rosenberg, D.W.; Giardina, C. Transcription factor NF-kappaB participates in the regulation of epithelial cell turnover in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G1282–G1291. [Google Scholar] [CrossRef]
- Place, R.F.; Noonan, E.J.; Giardina, C. HDAC inhibition prevents NF-kappaB activation by suppressing proteasome activity: Downregulation of proteasome subunit expression stabilizes I kappa B alpha. Biochem. Pharmacol. 2005, 70, 394–406. [Google Scholar] [CrossRef]
- Mitic, L.L.; Van Itallie, C.M.; Anderson, J.M. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: Lessons from mutant animals and proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G250–G254. [Google Scholar] [CrossRef]
- Smirnov, A.; Perez, R.; Amit-Romach, E.; Sklan, D.; Uni, Z. Mucin dynamics and microbial populations in chicken small intestine are changed by dietary probiotic and antibiotic growth promoter supplementation. J. Nutr. 2005, 135, 187–192. [Google Scholar] [CrossRef]
- Yang, X.; Yin, F.; Yang, Y.; Lepp, D.; Yu, H.; Ruan, Z.; Yang, C.; Yin, Y.; Hou, Y.; Leeson, S.; et al. Dietary butyrate glycerides modulate intestinal microbiota composition and serum metabolites in broilers. Sci. Rep. 2018, 8, 4940. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Martín, R.; Rossi, O.; Bermúdez-Humarán, L.G.; Chatel, J.M.; Sokol, H.; Thomas, M.; Wells, J.M.; Langella, P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.; Yitbarek, A.; Snyder, R.; Patterson, R.; Barta, J.R.; Karrow, N.; Kiarie, E. Responses of broiler chickens to Eimeria challenge when fed a nucleotide-rich yeast extract. Poult. Sci. 2019, 98, 1622–1633. [Google Scholar] [CrossRef]
- Liu, C.; Finegold, S.M.; Song, Y.; Lawson, P.A. Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov and description of Blautia wexlerae sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2008, 58, 1896–1902. [Google Scholar] [CrossRef]
- Liu, X.; Mao, B.; Gu, J.; Wu, J.; Cui, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Blautia—A new functional genus with potential probiotic properties? Gut Microbes 2021, 13, e1875796. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Borewicz, K.; White, B.A.; Singer, R.S.; Sreevatsan, S.; Tu, Z.J.; Isaacson, R.E. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 2011, 153, 124–133. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, H.; Xu, X.; Li, C.; Zhou, G. The gut microbiota in young and middle-aged rats showed different responses to chicken protein in their diet. BMC Microbiol. 2016, 16, 281. [Google Scholar] [CrossRef]
- Xi, Y.; Shuling, N.; Kunyuan, T.; Qiuyang, Z.; Hewen, D.; ChenCheng, G.; Tianhe, Y.; Liancheng, L.; Xin, F. Characteristics of the intestinal flora of specific pathogen free chickens with age. Microb. Pathog. 2019, 132, 325–334. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Q.; Huang, Z.; Lv, L.; Liu, X.; Yin, C.; Yan, H.; Yuan, J. Effect of Bacillus subtilis CGMCC 1.1086 on the growth performance and intestinal microbiota of broilers. J. Appl. Microbiol. 2016, 120, 195–204. [Google Scholar] [CrossRef]
- Banerjee, S.; Sar, A.; Misra, A.; Pal, S.; Chakraborty, A.; Dam, B. Increased productivity in poultry birds by sub-lethal dose of antibiotics is arbitrated by selective enrichment of gut microbiota, particularly short-chain fatty acid producers. Microbiology 2018, 164, 142–153. [Google Scholar] [CrossRef]
- Valles, C.; Mailhe, M.; Ricaboni, D.; Armstrong, N.; Alibar, S.; Vitton, V.; Lagier, J.-C.; Raoult, D.; Tidjani Alou, M. Negativibacillus massiliensis gen. nov., sp. nov., a new bacterial genus isolated from a human left colon sample. Microbiol. Res. 2021, 12, 29–42. [Google Scholar] [CrossRef]
- Wang, X.; Ding, Y.; Zhang, X.; Feng, Y.; Li, C.; Ge, Y.; Yang, Y.; Su, J.; Chu, X. The effects of degraded polysaccharides from Acanthopanax senticosus on growth, antioxidant and immune effects in broiler chicks based on intestinal flora. Poult. Sci. 2025, 104, 104933. [Google Scholar] [CrossRef]
- Kim, C.C.; Lunken, G.R.; Kelly, W.J.; Patchett, M.L.; Jordens, Z.; Tannock, G.W.; Sims, I.M.; Bell, T.J.; Hedderley, D.; Henrissat, B.; et al. Genomic insights from Monoglobus pectinilyticus: A pectin-degrading specialist bacterium in the human colon. ISME J. 2019, 13, 1437–1456. [Google Scholar] [CrossRef] [PubMed]
- Yüksel, E.; Voragen, A.G.J.; Kort, R. The pectin metabolizing capacity of the human gut microbiota. Crit. Rev. Food Sci. Nutr. 2024; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, R.; Li, G.; Zeng, T.; Chen, L.; Xu, W.; Gu, T.; Tao, Z.; Du, X.; Lu, L. Microbial fermented feed affects flavor amino acids and yolk trimethylamine of duck eggs via cecal microbiota–yolk metabolites crosstalk. Food Chem. 2023, 430, 137008. [Google Scholar] [CrossRef]
- Martin, E.; Fallschissel, K.; Kämpfer, P.; Jäckel, U. Detection of Jeotgalicoccus spp. in poultry house air. Syst. Appl. Microbiol. 2010, 33, 188–192. [Google Scholar] [CrossRef]
- Glendinning, L.; McLachlan, G.; Vervelde, L. Age-related differences in the respiratory microbiota of chickens. PLoS ONE 2017, 12, e0188455. [Google Scholar] [CrossRef]
- Haberecht, S.; Bajagai, Y.S.; Moore, R.J.; Hao Van, T.T.; Stanley, D. Poultry feeds carry diverse microbial communities that influence chicken intestinal microbiota colonisation and maturation. AMB Expr. 2020, 10, 143. [Google Scholar] [CrossRef]
- Farkas, V.; Csitári, G.; Menyhárt, L.; Such, N.; Pál, L.; Husvéth, F.; Rawash, M.A.; Mezőlaki, Á.; Dublecz, K. Microbiota composition of mucosa and interactions between the microbes of the different gut segments could be a factor to modulate the growth rate of broiler chickens. Animals 2022, 12, 1296. [Google Scholar] [CrossRef] [PubMed]
- Kubasova, T.; Faldynova, M.; Crhanova, M.; Karasova, D.; Zeman, M.; Babak, V.; Rychlik, I. Succession, replacement, and modification of chicken litter microbiota. Appl. Environ. Microbiol. 2022, 88, e01809–e01822. [Google Scholar] [CrossRef] [PubMed]
- Volf, J.; Crhanova, M.; Karasova, D.; Faldynova, M.; Kubasova, T.; Seidlerova, Z.; Sebkova, A.; Zeman, M.; Juricova, H.; Matiasovicova, J.; et al. Eggshell and feed microbiota do not represent major sources of gut anaerobes for chickens in commercial production. Microorganisms 2021, 9, 1480. [Google Scholar] [CrossRef]
- Bindari, Y.R.; Moore, R.J.; Van, T.T.H.; Walkden-Brown, S.W.; Gerber, P.F. Microbial taxa in dust and excreta associated with the productive performance of commercial meat chicken flocks. Anim. Microbiome 2021, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.; Dias, M.; Cervantes, R.; Pena, P.; Santos, J.; Vasconcelos Pinto, M.; Viegas, C. One Health Approach to Tackle Microbial Contamination on Poultries -A Systematic Review. Toxics 2023, 11, 374. [Google Scholar] [CrossRef]
- Apajalahti, J.; Vienola, K. Interaction between chicken intestinal microbiota and protein digestion. Anim. Feed Sci. Technol. 2016, 221, 323–330. [Google Scholar] [CrossRef]
- Lyte, J.M.; Keane, J.; Eckenberger, J.; Anthony, N.; Shrestha, S.; Marasini, D.; Daniels, K.M.; Caputi, V.; Donoghue, A.M.; Lyte, M. Japanese quail (Coturnix japonica) as a novel model to study the relationship between the avian microbiome and microbial endocrinology-based host-microbe interactions. Microbiome 2021, 9, 38. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, D.d.S.; Moreira Filho, A.L.d.B.; Araújo, W.J.d.; Sales, G.F.C.; Silva, H.M.d.; Oliveira, T.J.d.; Sousa, A.V.d.; Oliveira, C.J.B.d.; Givisiez, P.E.N. Gut Microbiome Structural Dynamics in Japanese Quail Across Developmental Stages. Microbiol. Res. 2025, 16, 167. https://doi.org/10.3390/microbiolres16080167
Gomes DdS, Moreira Filho ALdB, Araújo WJd, Sales GFC, Silva HMd, Oliveira TJd, Sousa AVd, Oliveira CJBd, Givisiez PEN. Gut Microbiome Structural Dynamics in Japanese Quail Across Developmental Stages. Microbiology Research. 2025; 16(8):167. https://doi.org/10.3390/microbiolres16080167
Chicago/Turabian StyleGomes, Daniela da Silva, Alexandre Lemos de Barros Moreira Filho, Wydemberg José de Araújo, Gustavo Felipe Correia Sales, Hemilly Marques da Silva, Thalis José de Oliveira, Antonio Venício de Sousa, Celso José Bruno de Oliveira, and Patrícia Emília Naves Givisiez. 2025. "Gut Microbiome Structural Dynamics in Japanese Quail Across Developmental Stages" Microbiology Research 16, no. 8: 167. https://doi.org/10.3390/microbiolres16080167
APA StyleGomes, D. d. S., Moreira Filho, A. L. d. B., Araújo, W. J. d., Sales, G. F. C., Silva, H. M. d., Oliveira, T. J. d., Sousa, A. V. d., Oliveira, C. J. B. d., & Givisiez, P. E. N. (2025). Gut Microbiome Structural Dynamics in Japanese Quail Across Developmental Stages. Microbiology Research, 16(8), 167. https://doi.org/10.3390/microbiolres16080167