1. Introduction
Klebsiella pneumoniae (
K. pneumoniae) is a major pathogen responsible for healthcare and community-associated infections, including hospital-acquired pneumonia, bloodstream infections (BSIs), urinary tract infections (UTIs), and wound and soft tissue infections. This bacterium’s ability to develop resistance to multiple antibiotics presents a significant challenge for effective treatment [
1]. In particular,
K. pneumoniae strains producing extended-spectrum beta-lactamases (ESBLs) are associated with poor clinical outcomes [
2]. For example, clinical failure at 7 days was higher in ESBL-producing
K. pneumoniae UTIs compared to non-ESBL strains [
3]. Inadequate empirical antibiotic coverage was a major factor, with appropriate treatment achieved in only 37.7% of ESBL-producing cases, compared to 92.9% for non-ESBL cases [
3].
The World Health Organization (WHO) classifies both third-generation cephalosporin-resistant (3GCR) and carbapenem-resistant
K. pneumoniae as critical threats to human health [
4]. 3GCR
K. pneumoniae is particularly associated with increased in-hospital mortality, prolonged hospital stays, and higher healthcare costs [
4]. Mortality rates are notably high; the 30-day case fatality rate (CFR) for ESBL-producing
K. pneumoniae BSIs has been reported at 10.6% [
5]. In a separate study of 166 patients with BSIs, the overall mortality rate was 43.0% for those infected with ESBL-producing
K. pneumoniae [
6]. In 2019 alone, third-generation cephalosporin-resistant
K. pneumoniae accounted for approximately 50,100 deaths globally [
7].
The most widespread ESBL gene,
blaCTX-M-15, has been reported worldwide and is frequently linked to various
K. pneumoniae clonal lineages, including ST101 [
8], ST1427 [
9], ST307 [
10,
11], and ST394 [
12]. ESBL genes are commonly located on large, mobile plasmids that can be transferred between species.
K. pneumoniae serves as a key reservoir for antimicrobial resistance (AMR) genes, effectively acquiring and disseminating these plasmids within its own population and across other Enterobacteriaceae species.
Horizontal transfer of ESBL-encoding plasmids has been documented across several
K. pneumoniae sequence types (STs) and other Enterobacteriaceae species. A 120 kbp IncFIIK conjugative plasmid encoding
blaCTX-M-15, along with seven other AMR genes, was detected in eight isolates from four different
K. pneumoniae STs (ST416, ST321, ST280, and ST628) and
Enterobacter cloacae over a one-year surveillance study in a pediatric oncology department in the Czech Republic [
13].
In Tanzania, the horizontal transfer of an epidemic IncFII/IncR plasmid (pK012_2) carrying
blaCTX-M-15 was detected across 48 different
K. pneumoniae STs from stool samples collected from both healthy and hospitalized children [
14]. This widespread dissemination of a single plasmid across unrelated bacterial clones highlights how F-like conjugation modules contribute to efficient horizontal gene transfer with minimal fitness costs.
In 2018, a clonal outbreak of
K. pneumoniae ST628 was recorded in a UK district general hospital [
15]. The outbreak strain harbored a multi-drug resistant (MDR) plasmid, pESBL-PH encoding 11 AMR genes:
aac(3)-IIe,
aac(6′)-Ib-cr,
aph(3″)-Ib,
aph(6)-Id,
blaCTX-M-15,
blaOXA-1,
blaTEM-1B,
dfrA14,
qnrB1,
sul2, and
tet(A). Antibiotic susceptibility testing (AST) confirmed this plasmid was responsible for resistance to six drug classes, including aminoglycosides, fluoroquinolones, cephalosporins, diaminopyrimidines, sulfonamides, and tetracyclines [
15]. Here, we aimed to determine whether the plasmid pESBL-PH-2018, first identified on 3 March 2018 from a UTI patient, was present in other
K. pneumoniae sequence types (STs). We sought to identify structurally similar plasmids within the complete plasmid database downloaded from NCBI, using stringent thresholds.
3. Results
3.1. Plasmid Similarity Against pESBL-PH-2018: An Overview
The strict search criteria yielded 61 similar plasmids. The similarity between the query plasmids and the reference pESBL-PH-2018 was compared using a series of methods. The Mash distance metric, based on the Jaccard index of
k-mer sketches (subsequences of length
k) was initially used to assess plasmid similarity (
Supplementary File S1). A mean Mash similarity (1—Mash distance) of 0.9992 was recorded, with a standard deviation (σ) of 0.0007 and an interquartile range (IQR) of 0.9988–0.999. Additionally, the number of shared hashes was assessed as a ratio. Across the collection of 61 plasmids samples, the hash ratio ranged from 0.902 to 1.00 (mean: 0.968, IQR: 0.954–0.998, median: 0.969, σ: 0.029).
The nucleotide coverage and identity were also compared between each of the 61 query plasmids and the reference, pESBL-PH-2018. BLASTn (version 2.14.1) coverage for the plasmids against pESBL-PH-2018 ranged from 91 to 100% (mean: 97.82%, IQR: 97–100%, median: 98.0%, σ: 2.34). The BLASTn identity of the query plasmids against pESBL-PH-2018 was high; range 99.87–100.00% (mean: 99.99%, IQR: 99.99–100.00%, σ: 0.019). Finally, the length of the plasmids was assessed. The 61 query plasmids had a mean length of 244,365 bp. When each plasmid was compared against the reference length of pESBL-PH-2018, 247, 179 bp, the IQR for plasmid length was between 97.56–99.99% of the length of pESBL-PH-2018. Taken together, the high Mash similarity, shared hashes, high coverage, and nucleotide identity, combined with a similar length, suggest that all 61 plasmids are highly similar to pESBL-PH-2018.
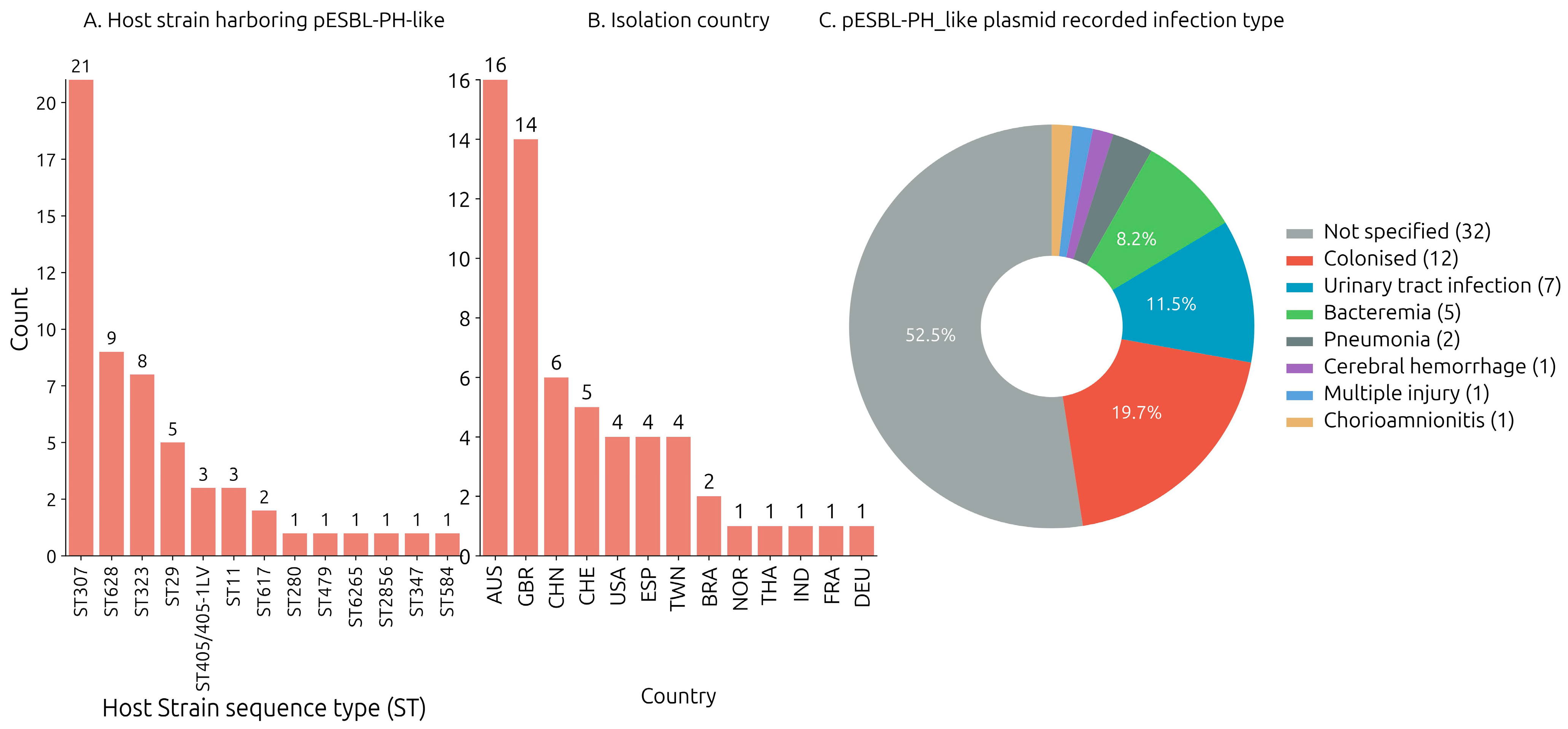
3.2. Identification of pESBL-PH-2018-like Plasmids: Host Strain and Isolation Source
The host bacterium strain harboring each of the query plasmids were downloaded from the NCBI nucleotide database. Across the collection of 57 plasmid samples where a host bacterium ST designation was assigned, 13 unique sequence types (STs) were identified. The pESBL-PH-2018-like plasmid was detected most frequently in
K. pneumoniae ST307 (
n = 21/57, 36.84%). Furthermore, pESBL-PH-2018-like plasmids were found in five STs, where the ST was identified three or more times: ST628 (
n = 9/57, 15.78%), ST323 (
n = 8/57, 14.03%), ST29 (
n = 5/57, 8.77%), ST405 (
n = 3/57, 5.26%), and ST11 (
n = 3/57, 5.26%). In addition, the pESBL-PH-2018-like plasmid was identified in seven other STs: ST617, -280, -479, -6265, -2856, and ST584, respectively (
Figure 1A). Across 60 samples where a host strain species was recorded, the pESBL-PH-2018 like plasmid was predominately detected in
K. pneumoniae (
n = 57/60, 95%). Separately, three similar plasmids were detected in two different genera, while a single plasmid was detected in different species: plasmid P1 was detected in a
Klebsiella oxytoca strain; plasmid 2, in KPN029
Klebsiella variicola ST347; and pCUVET16-321.1, in an
Escherichia coli ST479 strain. Together, these results suggest different
Klebsiella genera and other species may be receptive carriers for pESBL-PH-2018-like plasmids.
The pESBL-PH-2018-like plasmid was identified in 13 unique countries between 2012 and 2023 (
Figure 1B). The available Biosample data from NCBI indicated the plasmid (including pESBL-PH-2018) was most often found in Australia (26.22%,
n = 16/60), the United Kingdom (22.95%,
n = 14/60), and China (9.83%,
n = 6/60). Separately, the plasmid was identified ≥4 times in Switzerland, USA, Spain, and Taiwan (
Figure 1B). Available Biosample data also indicated the pESBL-PH-2018-like plasmids (including pESBL-PH-2018) were identified from a range of infection types including urinary tract infections (11.3%,
n = 7), bacteremia (8.1%,
n = 5), pneumonia (3.2%,
n = 2), cerebral hemorrhage (1.6%,
n = 1), acquisition from wounds via injuries (1.6%,
n = 1), and chorioamnionitis (1.6%,
n = 1), respectively (
Figure 1C).
Interestingly, among the 61 query plasmids similar to pESBL-PH-2018, the majority,
n = 57/61 (93.44%), were identified from human hosts. Forty-seven of these samples had a clinical source isolation (
Supplementary File S1); the remaining 10 samples lacked data on their isolation source. In hospital settings, high and frequent use of antibiotics can create strong selective pressure favoring bacteria that carry MDR plasmids.
Four samples, however, n = 4/61 (6.55%), were identified from the environment and/or non-human hosts, including one sample collected from stream water (pEW1149-1), one sample from river water (pEW758-CTX-M-15), another sample from milk in an environmental sample (pKP102-1), while the pESBL-PH-2018-like plasmid pCUVET16-321.1, was identified in a urine sample from a ST479 E. coli sample obtained from a dog. These results suggest that the pESBL-PH-2018-like plasmid can be found beyond the confines of clinical settings.
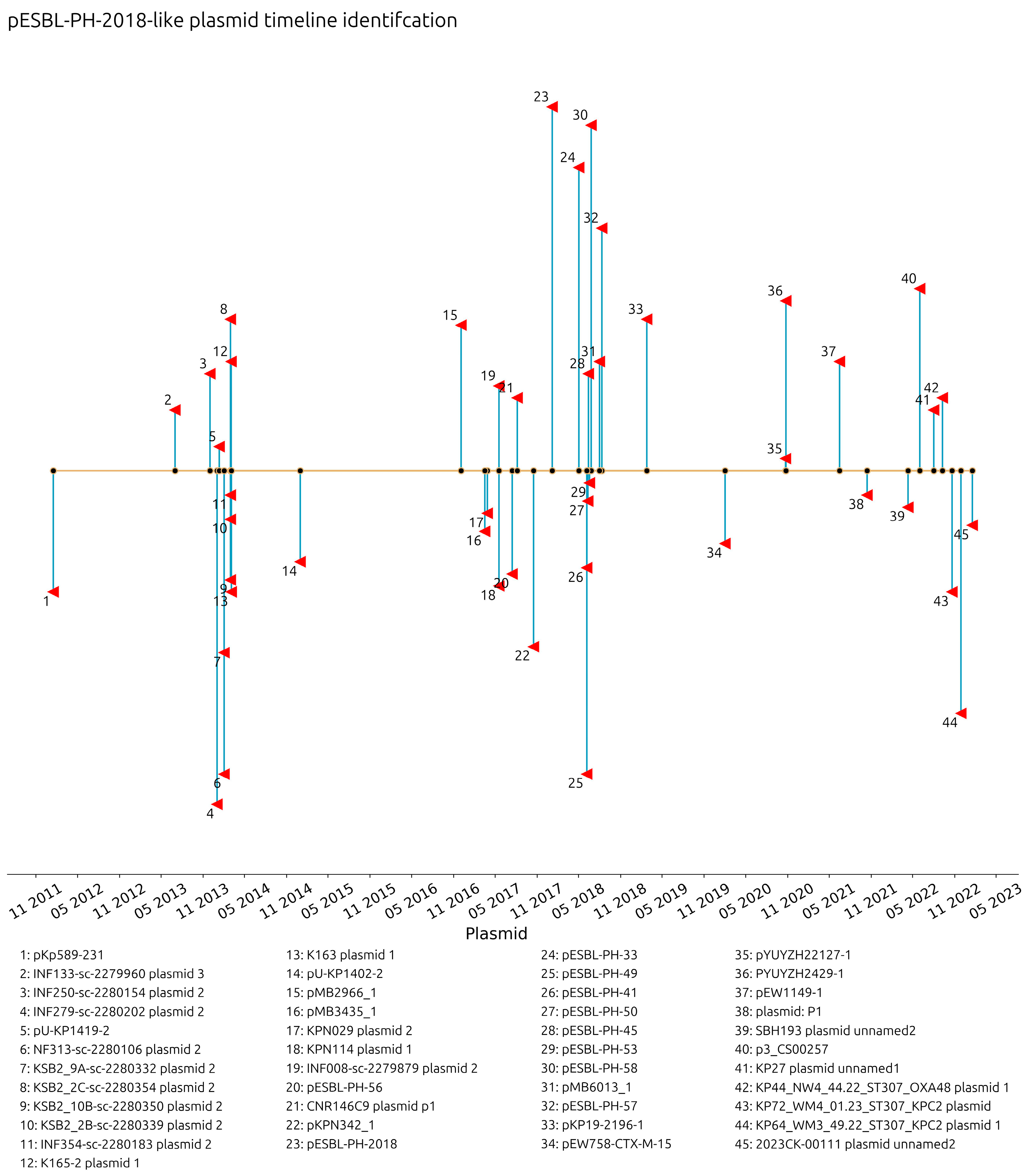
The collection dates of plasmids with a recorded year, month, and day were investigated. Plasmids similar to pESBL-PH-2018 were collected from 16 January 2012 in Barcelona, Spain (pKp589-231, accession: CP028817.1) through to 17 January 2023 in the USA (2023CK-0011, plasmid unnamed, accession: CP131947.1). The interval between the detection of these two plasmids is 11 years and 4 days. Similar plasmids were detected from 25% (
n = 11) of the samples with a collection date in 2014, while 20.45% (
n = 9) were collected in 2018; however, ≥3 similar plasmids were recorded in 2014, 2017, 2018, 2020, and 2022 (
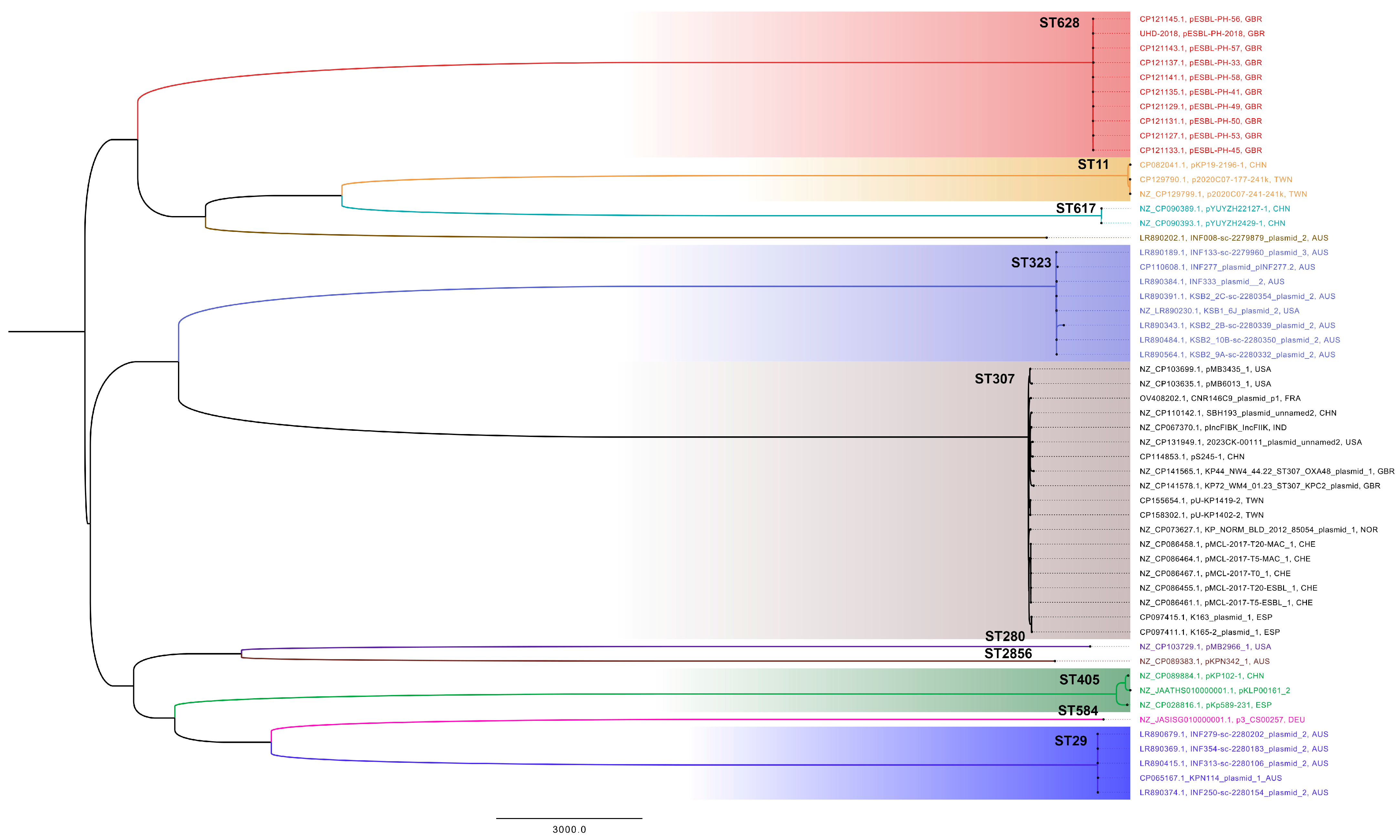
Figure 2). These results reveal a consistent presence of ESBL-encoding plasmids similar to pESBL-PH-2018. A recombination-filtered phylogenetic tree of host bacterial strains revealed that the plasmids were carried by distinct bacterial clones. Across these clones, evidence of expansion of the host strain with the plasmid is seen (
Figure 3).
3.3. Single Nucleotide Polymorphism Analysis
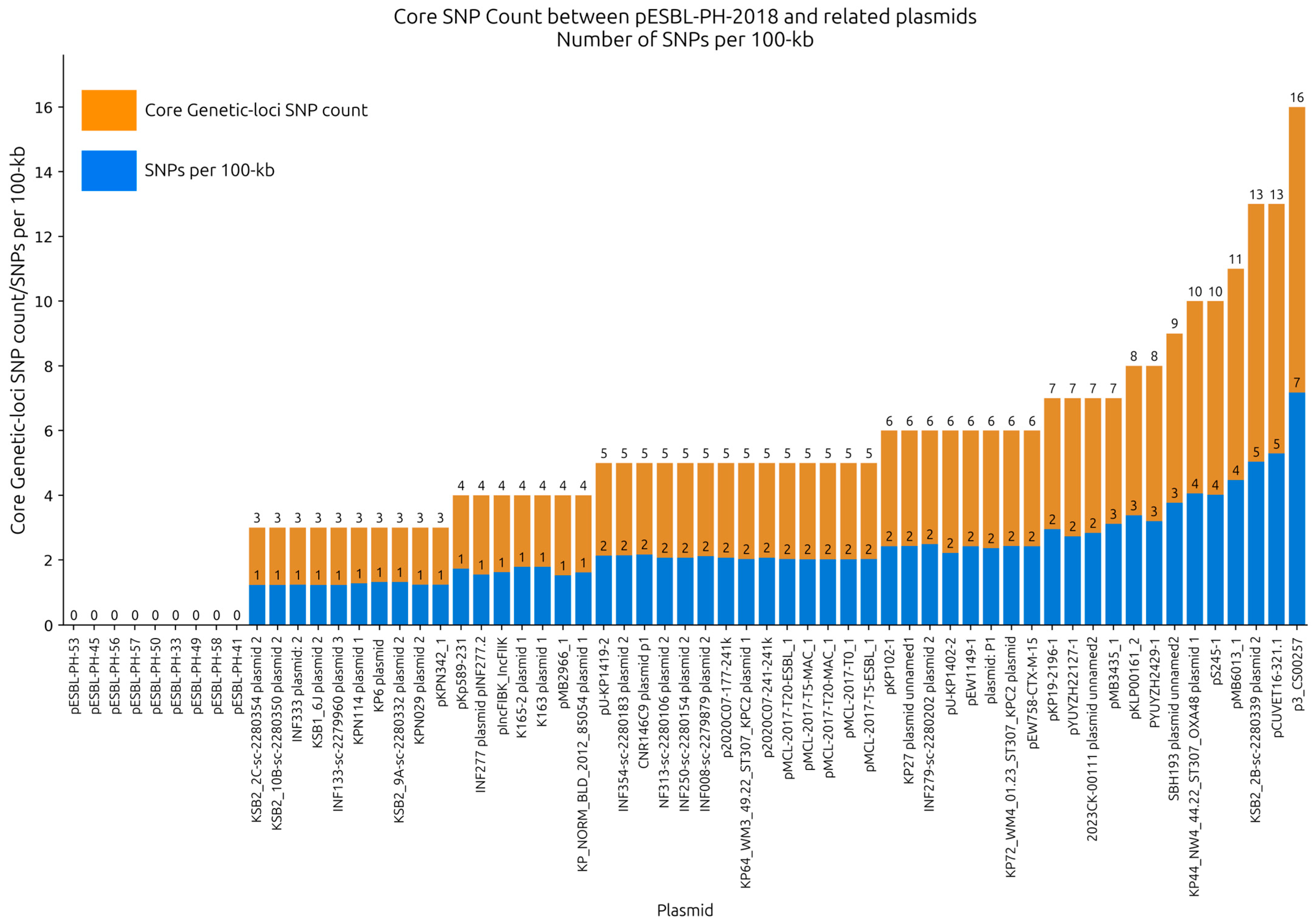
Single nucleotide polymorphisms (SNPs) between pESBL-PH-2018 and the query plasmids were investigated to provide high-resolution analysis of their relatedness. SNP analysis was performed on recombinant-removed query plasmids. Across the selection of plasmids, an average of 4.95 SNPs were detected (range: 0–16, IQR: 3–6); 93.44% (
n = 57/61) of plasmids had an SNP count ≤10, while 65.5% (
n = 40/61) had an SNP count ≤ 5. With the exception of CNR146C9 plasmid 1 and p3_CS00257 each of which had a recombination region of 6757 bp and 17,782 bp, respectively, a mean recombinant region of 531 bp was identified (range: 0–2396 bp), which, on average, only constituted 0.22% of their overall plasmid length, revealing that the plasmids shared large stretches of identical sequences. Across all plasmid samples, an average of 1.95 SNPs/100 kb was identified, suggesting that the plasmids were closely related to pESBL-PH-2018 (
Figure 4,
Supplementary File S1).
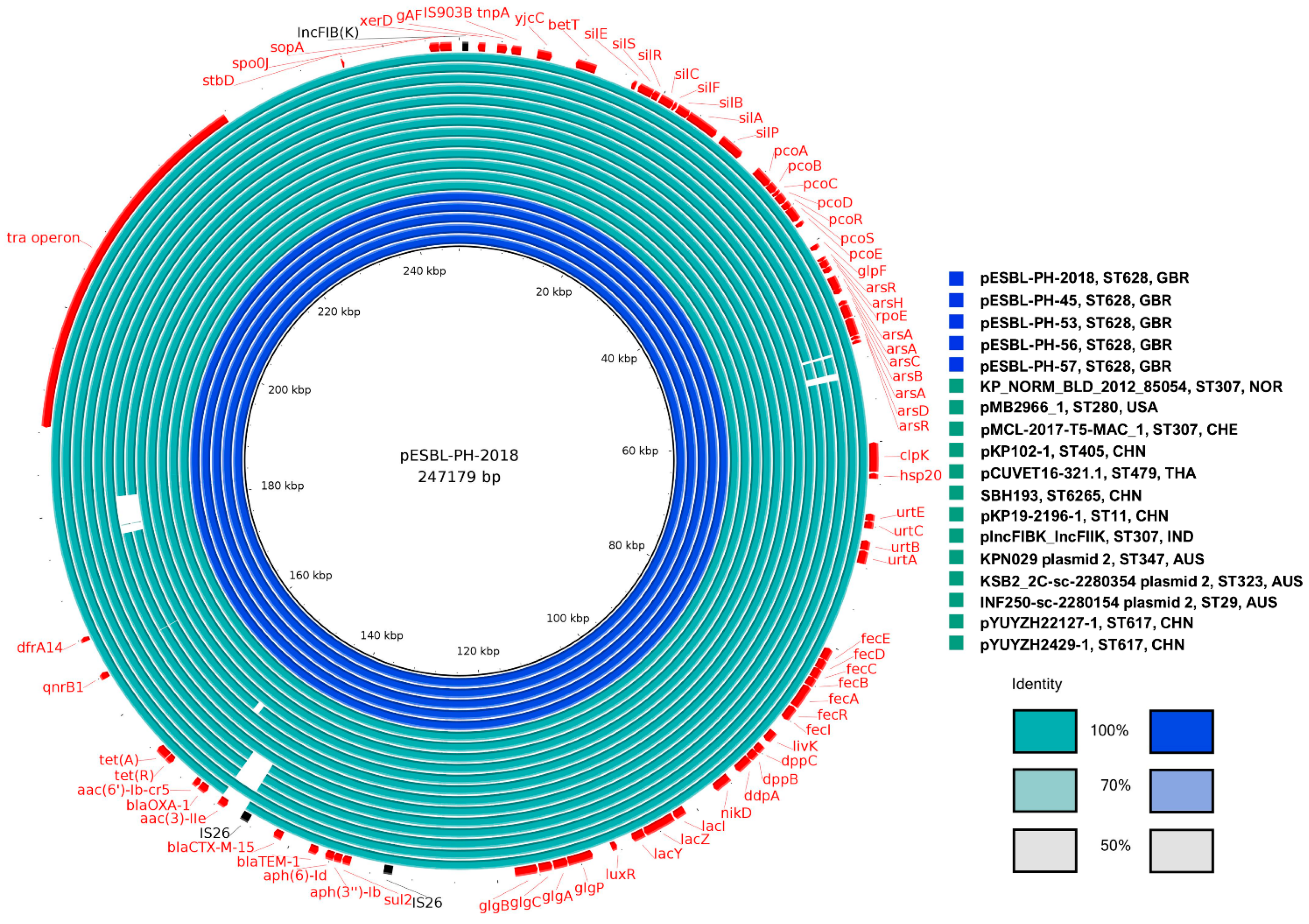
3.4. Core Genome Analysis
The core genome analysis was performed on 12 plasmids (
Table 1) from
K. pneumoniae strains, representing 12 unique sequence types (STs). This approach aimed to identify conserved genetic elements constituting the plasmid backbone, which are essential for plasmid maintenance, stability, and potential conjugative transfer across diverse host strains. A total of 210 genes were shared across all plasmids, with a combined nucleotide length of 182,216 bp. Additionally, 90.9% of genes (
n = 281/309) were classified as either core genes (present in 99–100% of plasmids) or shell genes (present in 15–95% of plasmids). Only 9.1% of genes (28/309) were considered cloud or accessory genes, highlighting the substantial conservation of core genetic elements across diverse host strains. The core backbone and shared gene content are visualized for representative STs in
Figure 5.
While the plasmid analysis initially identified 13 unique host strain STs, the plasmid pMB2966_1 from K. pneumoniae ST280 was excluded from the core genome analysis due to the presence of a segmental duplication. This duplication encompassed several AMR genes, including sul2, aph(3″)-Ib, aph(6)-Id, blaTEM-1, and blaCTX-M-15. Including this plasmid in the analysis would have skewed the results by artificially inflating the number of genes classified as core, thereby distorting the overall structure of the core genome.
The exclusion of pMB2966_1 ensured that the identified core genome reflected genuinely conserved elements, rather than artifacts arising from gene duplication or rearrangement. Interestingly, despite differences in AMR gene content, all plasmids shared several stress-related genes with 100% identity and coverage, including silver (silS, silR, silC), copper (pcoABCR), and arsenic (arsABCR) heavy metal resistance genes, alongside the heat shock gene hsp20. Furthermore, all plasmids were classified as IncFIB(K) and were predicted to be conjugative, possessing essential components for horizontal gene transfer, such as a relaxase, T4SS, and T4CP.
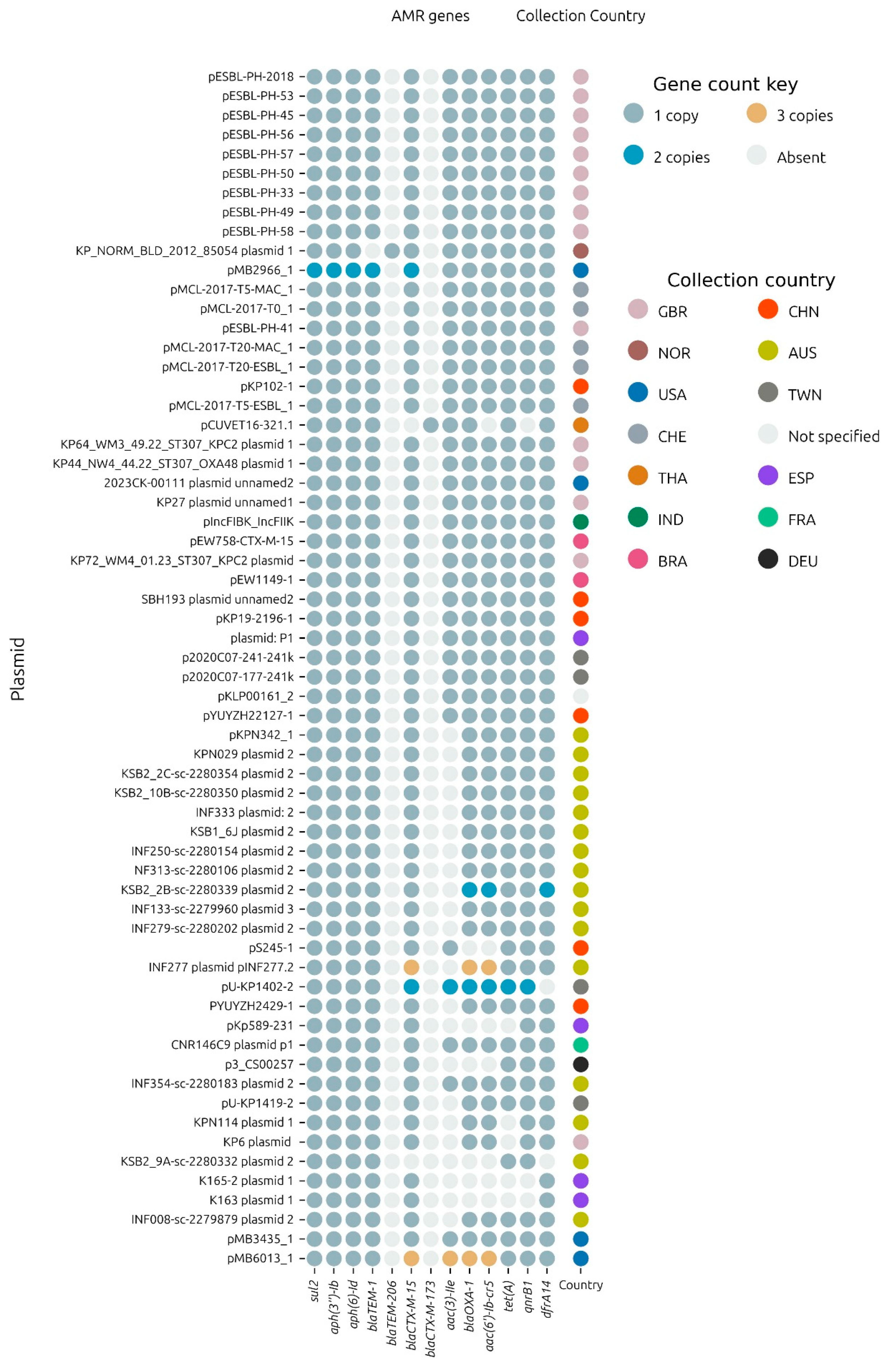
3.5. AMR Variants
From the collection of plasmids including pESBL-PH-2018, 54.8% (
n = 34/62) carried exactly one copy of the 11 AMR genes:
aph(3″)-Ib,
aph(6)-Id,
blaTEM-1B,
blaCTX-M-15,
aac(3)-IIe,
blaOXA-1,
aac(6′)-Ib-cr,
tet(A),
qnrB1,
dfrA14 and
sul2. Plasmids pMB2966_1 and pMB6013_1 carried multiple copies of these genes, including either 2 or 3 copies each of the ESBL gene,
blaCTX-M-15, respectively (
Figure 6). In total, 58% of the plasmids carried either one or more of the total complement of 11 AMR genes carried by pESBL-PH-2018. Notably, a common variant, which lacked only
aac(3)-IIe was detected in 24.19% (
n = 15/62) of samples. From these 15 samples, 13 samples were identified from Australia, confirming that the
aac(3)-IIe aminoglycoside gene was commonly missing from plasmids related to pESBL-PH-2018 sourced from Australia.
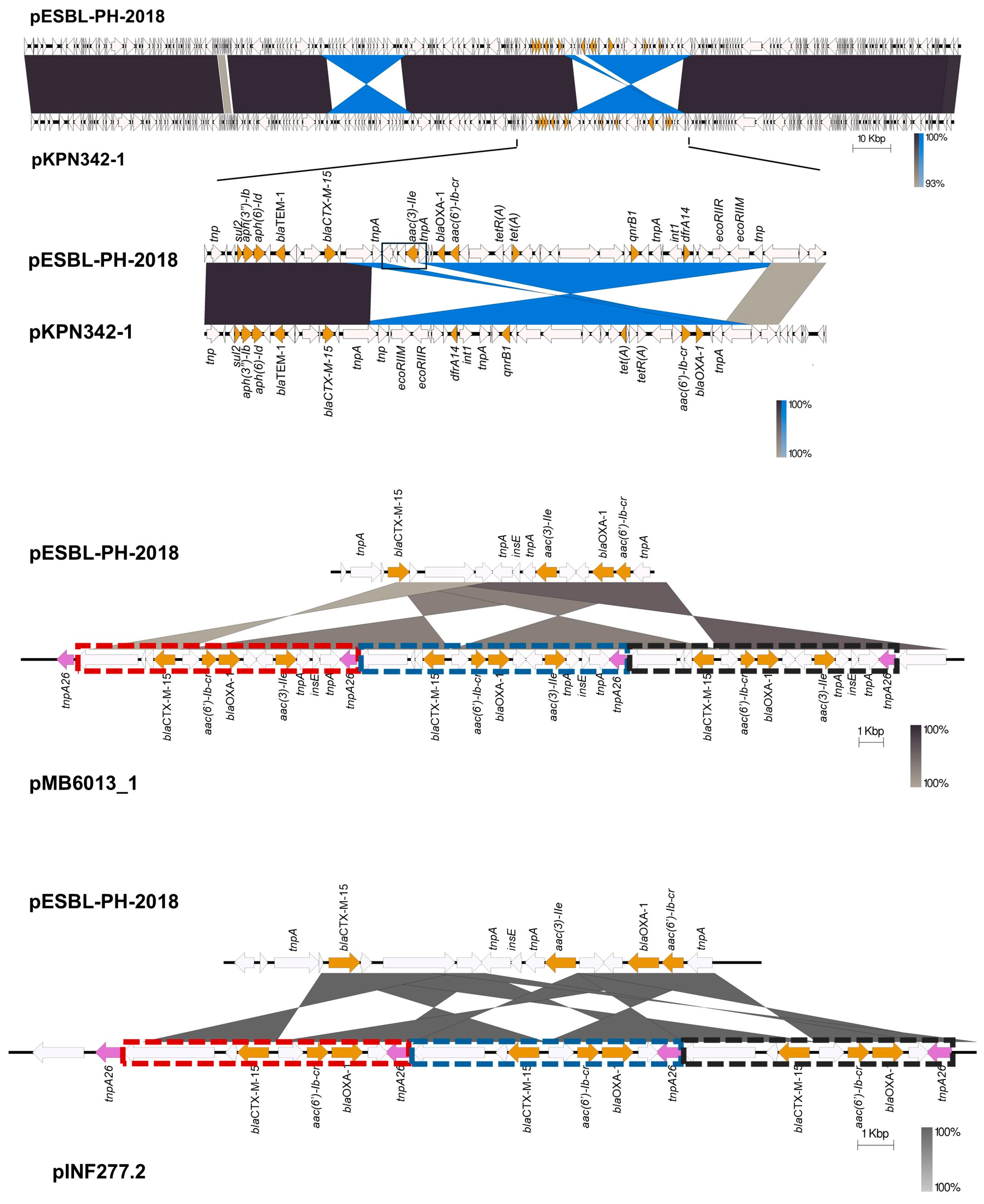
Although similar plasmids to pESBL-PH-2018 were detected, a number of variants with different numbers of the same AMR genes were observed. The aminoglycoside resistance gene,
aac(3)-IIe, was absent from a number of plasmids. In comparison to pESBL-PH-2018, plasmid pKPN342_1 (accession: CP089384.1), an AMR variant lacking
aac(3)-IIe, had a deletion of 3420 bp, including
aac(3)-IIe and a complete copy of IS
26 located between two inverted sequences (
Figure 6). A similar deletion of 2600 bp encompassing
aac(3)-IIe was identified in plasmid KBS2_10B (accession: LR8900485.1). However, in this plasmid, both copies of the IS
26 between
aac(3)-IIe were retained.
Separately, KP_NORM_BLD_2012_85054 plasmid 1 (accession: CP073628.1) from K. pneumoniae ST307 encoded the TEM variant, blaTEM-206. This variant was due to an SNP at nucleotide position 25, resulting in a missense variant, whereby the non-polar amino acid alanine was replaced by the polar amino acid threonine at amino acid position 9.
Variants with two copies of several AMR genes, including the ESBL gene, blaCTX-M-15, were identified. pMB2966_1 had two copies each of the following genes: sul2, aph(3″)-Ib, aph(6)-Id, blaTEM-1, and blaCTX-M-15, respectively. In pESBL-PH-2018, these AMR genes are present within a 15,310 bp IS26 pseudo-compound transposon (PCT). However, in pMB2966_1, a segmental duplication (mobilization to another genomic context) of a 16,374 bp region, including an inverted copy, carrying these genes was observed. Both regions carrying these AMR genes shared 12,212 bp with the IS26 PCT from pESBL-PH-2018, but both segments lacked IS26 insertion sequences bracketing the 5 AMR genes, confirming that the duplication of these genes did not occur due to an IS26-mediated translocatable unit (TU) amplification. Additionally, plasmid 2 (accession: LR890344.1) from K. pneumoniae ST323, obtained from a rectal swab, had a segmental duplication of 10.98 kb, including the AMR gene, dfrA14. Similar to pMB2966_1, the second segmental copy lacked IS26 elements on either side of the duplication.
For plasmid pU-KP1402-2, two copies each of blaCTX-M-15, aac(3)-IIe, blaOXA-1, aac(6′)-Ib-cr, tet(A), and qnrB1 were identified. Here, blaCTX-M-15 and aac(3)-IIe were duplicated as an additional inverted copy associated with a single copy of IS26 tnpA26. Another inverted duplication was also observed for blaOXA-1 and aac(6′)-Ib-cr, with both copies having inwardly facing tnpA26 transposases. Interestingly, both tet(A) and qnrB1 were also duplicated.
Two plasmid variants, pMB6013_1 (accession: CP103636.1) and pINF277.2 (accession: CP110610.1), with a copy number of three for the ESBL gene
blaCTX-M-15 and other AMR genes were also investigated. Here, IS
26-TU tandem amplification was identified. While pESBL-PH-2018 was similar to pMB6013_1, the AMR region varied. Relative to pESBL-PH-2018, a transposase gene and the ESBL gene
blaCTX-M-15 are inverted. This inversion lies upstream of an IS
26 tnpA26 and downstream of an inverted region carrying three AMR genes,
aac(6′)-Ib-cr,
blaOXA-1, and
aac(3)-IIe. Together, this forms a section of four AMR genes bracketed by IS
26 tnpA26 on each side (
Figure 7). This region was duplicated, generating three tandem copies of these AMR genes. The AMR tandem repeat is 11,009 bp, measured from the inverted repeat left (IRL) of IS
26 to the shared region of the Tn3 transposase fragment (marked boxes
Figure 7). The genomic organization indicates IS
26-mediated TU amplification may have created the tandem array.
Separately, pINF277.2 from
K. pneumoniae ST323, isolated from a bronchoalveolar lavage (BAL) sample from a patient with pneumonia, also had a similar IS
26-mediated TU amplification for the three AMR genes:
blaCTX-M-15,
aac(6′)-Ib-cr, and
blaOXA-1. Relative to pESBL-PH-2018,
blaCTX-M-15 is inverted and lies upstream of a shared IS
26 tnpA26 transposase. The AMR region from pESBL-PH-2018, including
aac(6′)-Ib-cr,
blaOXA-1 and
tnpA26, is inverted and located downstream of
blaCTX-M-15 (
Figure 7). This region in pINF277.2, bracketed by
tnpA26, is duplicated three times, increasing the copy number for each of these genes to three. The IS
26-mediated TU tandem array here was 7559 bp (marked boxes,
Figure 7).
3.6. Host Strain Background
The host strain background for 56 strains with a complete chromosome was investigated for AMR, virulence, and stress-associated factors. In particular, gene mutations associated with carbapenem resistance were analyzed. The pESBL-PH-2018-like plasmid was present in a background associated with carbapenem resistance mechanisms. Notably, 26.78% (
n = 15/56) of the strains encoded a carbapenem resistance gene on an accessory plasmid in addition to carrying a pESBL-PH-2018-like plasmid (
Figure 8). In addition, 17.85% (
n = 10/56) of the strains had a truncation in the outer membrane porin gene,
ompK36, disruptions of which can reduce the permeability of the outer membrane to carbapenems. Porin truncations were present in four strains, which also carried a carbapenemase gene on an accessory plasmid (
Figure 8). Three strains carried the
ompK36 variant,
ompK36_D135DGD, associated with porin constriction mediated by the di-amino acid (Gly115-Asp116) insertion into loop 3 of the OmpK36 porin [
32]. This change has been associated with restricted access to carbapenems into the host cell. Two of these variants were found, in addition to carbapenem resistance genes present on separate plasmids (
Figure 8,
Supplementary File S2).
Colistin resistance associated gene variants were also detected in 14.28% (n=8/56) strains. Here, 5 strains carried the pmrB gene variant: pmrB_R256G, 1 carried the pmrB_Y358N variant, while 2 strains carried both the pmrB_T157P alongside a gene truncation in mgrB. Such changes have been associated with constitutive activation of the two-component system, PmrA-PmrB, leading to a reduction in the negative charge of the bacterial lipopolysaccharide (LPS) present on the bacterial outer membrane. The reduction in negative charge decreases the binding affinity of colistin (a cationic peptide), leading to colistin resistance.
Notably, a large proportion, 92.85% (n = 52/56), of the isolates were predicted to encode the chromosomal fosfomycin resistance gene, fosA. In addition, a number of predicted quinoline resistance-inducing mutations impacting either DNA gyrase or topoisomerase IV were detected. In particular, all representative ST11 and ST307 isolates encoded the gyrA S83I and parC S80I double mutations. A proportion of strains, 14.28% (n = 8/56), also encoded the virulence-associated YbtPQ ABC transporter, a key transporter system involved in both iron acquisition during infection and broad export of antimicrobials. Taken together, AMR, virulence and stress-associated typing of the chromosomes and plasmids co-present with the pESBL-PH-2018-like plasmid reveal a genomic background associated with genes conferring resistance to critical and last-resort antimicrobials, such as carbapenems and colistin, in addition to the six classes of AMR resistance conferred by pESBL-PH-2018.
4. Discussion
Multiple lines of evidence demonstrate the widespread dissemination of an pESBL-PH-2018-like plasmid. These include a high Mash similarity, high coverage and nucleotide identity, and a low SNP difference in the core genetic loci of query plasmids. Similar plasmids also carried either identical or a similar number of the same AMR genes, alongside an identical plasmid replicon, heavy metal resistance genes, and conjugative transfer genes. The broad-host range of CTX-M-15-encoding plasmids has been previously recorded [
11,
14]. In addition, expansion of different bacterial clones harboring a similar CTX-M-15 encoding plasmid has been reported [
33,
34]. pESBL-PH-2018 may represent another plasmid, which can transfer between hosts. Once transferred, clonal expansion within receptive hosts can occur.
Plasmids similar to pESBL-PH-2018 were identified from 2012 to 2023, further confirming the long-term persistence of a pESBL-PH-2018-like plasmid. High conjugation efficiency has previously been reported for CTX-M-15-encoding plasmids, with high conjugation frequencies in the order of 10
−1 recorded [
35]. Together, the presence of similar plasmids in distinct hosts, alongside the identification of conjugative transfer genes, may indicate that horizontal gene transfer (HGT) could be responsible for the widespread dissemination of a pESBL-PH-2018-like plasmid. However, separate conjugation assays are required to determine the conjugative frequency for pESBL-PH-2018 and similar plasmids. Plasmid transfer efficiency can be highly variable depending on numerous factors, including the recipient strain, environmental conditions, selective pressures, and plasmid compatibility. These factors should be explored in future work.
The AMR region varied between the plasmids. Notably, the aminoglycoside resistance gene
aac(3)-IIe was absent on a number of plasmids and may have been lost via IS
26-mediated deletion, as the strains lacked both the resistance gene and IS
26 (such as pKPN342-1). For two plasmids, pMB6013_1 and pINF277.2, the copy number of the ESBL gene
blaCTX-M-15, alongside other AMR genes, was repeated three times in a tandem array. Rolling Circle Replication (RCR) involves the amplification of DNA through the continuous replication of a translocatable unit (TU) formed by IS
26. IS
26 mediates the excision of a DNA segment containing resistance genes, generating a circular TU. During RCR, DNA polymerase initiates replication at a nicked site, producing a long single-stranded DNA (ssDNA) tail, which is later converted into double-stranded DNA (dsDNA). Multiple copies of the TU are created rapidly, and IS
26 facilitates their reintegration into the plasmid, resulting in the tandem duplication of resistance genes. For plasmids pINF277.1 and pMB6013_1, IS
26-mediated TU formation and RCR may have contributed to the amplification of resistance genes including
blaCTX-M-15, enhancing resistance and promoting their persistence and dissemination. Tobramycin selection pressure has been shown to lead to the duplication of the
aphA1 kanamycin and neomycin resistance gene from an
Acinetobacter baumannii clinical strain MRSN56, due to IS
26-mediated TU formation [
36]. The AMR region of pESBL-PH-2018 may be subject to rearrangements and resistance gene duplications due to the presence of IS
26, combined with antibiotic selection pressure, which is often encountered in clinical settings. Concerningly, for both plasmids with tandem repeats of AMR genes, dual β-lactamase-encoding genes,
blaCTX-M-15 and
blaOXA-1, were identified. IS
26-mediated co-amplification of two β-lactamase-encoding genes can yield a phenotype which is often non-susceptible to carbapenems, such as meropenem or ertapenem [
37].
Beyond the presence of AMR and virulence genes present on the pESBL-PH-2018-like plasmid, the genotypic background of these strains was investigated to identify additional resistant/virulence-linked determinants associated with carbapenem and colistin resistance. A proportion of the strains carried the chromosomal integrative and conjugative element ICE
Kp encoding the ABC transporter YbtPQ. Recently, the YbtPQ transporter has been shown to reduce bacterial susceptibility to several classes of antibiotics, including carbapenems, likely mediated via broad range antimicrobial export [
38]. The co-presence of carbapenemase genes in 7/8 of the strains encoding YbtPQ may increase the minimum inhibitory concentration (MIC) for carbapenems and other classes of antibiotics while contributing towards enhanced virulence via iron acquisition.
Furthermore, disruptions in the outer membrane porin gene
ompK36, in concert with the presence of β-lactamase genes, have been linked with the development of non-carbapenemase-producing carbapenem-resistant Enterobacterales (non-CP-CRE). For example, a non-CP-CRE
K. pneumoniae isolate, MB101, carried tandem amplification of a TU carrying
blaOXA-1, together with an IS
Ecp1 transposition unit inserted into
ompK36 [
39]. Six strains carried
ompK36 truncations without the presence of either chromosomal or plasmid carbapenem resistant genes. Notably, in two strains, multiple plasmid
blaCTX-M-15 genes were detected with either
ompK36 truncations (strain 3221, chromosomal accession: NZ_CP103635.1) or
ompK35 (strain U-KP1402, chromosomal accession: CP158302.1), yielding a potential non-CPE genotype. These two strains, and the other four strains with a single
blaCTX-M-15 copy, may have a background primed for the development of carbapenem resistance, a worrying scenario, given the presence of CTX determinants, which already confer resistance to cephalosporins, and other strains which already carried carbapenem resistant genes on separate plasmids. Such strains will be harder to treat and may present more opportunities for onward ESBL plasmid dissemination.
Low levels of antibiotics or heavy metals in polluted environments, or resulting from antibiotic consumption, can promote the selection and enrichment of MDR plasmid carriage among bacteria. Notably, very low concentrations of individual antibiotics, heavy metals, or their combinations can select for the large 220 kbp MDR plasmid, pUUH239.2, which confers resistance to aminoglycosides, β-lactams, tetracycline, macrolides, trimethoprim, sulfonamide, silver, copper, and arsenic [
40]. Both antibiotics and heavy metals at sub-minimum inhibitory concentrations (sub-MICs) can select for pUUH239.2. For instance, the minimum selective concentration (MSC) for tetracycline is 17-fold lower, while for arsenite, it is approximately 140-fold lower than the corresponding MICs [
40].
Areas affected by human pharmaceutical use may promote the maintenance and enrichment of MDR plasmids, contributing to their persistence and dissemination. This is particularly concerning for ESBL-encoding plasmids such as pESBL-PH-2018-like plasmids, which often encode resistance to both antimicrobials and heavy metals. In European surface waters, trimethoprim has been detected at concentrations reaching several hundred ng/L [
41]—levels that exceed the MSC for trimethoprim.
While antibiotic and heavy metals may contribute towards the selection of MDR plasmids, non-native strains harboring MDR plasmids may be outcompeted in environmental settings. MDR plasmids are infrequently found in environmental settings such as stream and river water due to the competitive exclusion exerted by the native microbiota. A recent study demonstrated that both clinical and environmental ST147
K. pneumoniae strains carrying
blaKPC-3 on an IncN plasmid rapidly declined in urban runoff water due to competition from indigenous microbial communities [
42]. The abundance of the
blaKPC-3 gene decreased significantly over time, correlating with the loss of cultivable
K. pneumoniae rather than gene loss, indicating that host cell death was the primary cause of reduction. Additionally, the rise in Alphaproteobacteria abundance over time suggested that these bacteria may be particularly effective competitors against invasive species. In contrast, when incubated in sterile ultra-pure water (UP), where native microbiota were absent, the ST147
K. pneumoniae strains maintained viability and plasmid stability over eight days [
42]. This highlights that in natural environments, where microbial diversity is high and resources are limited, non-native
K. pneumoniae carrying MDR plasmids, such as pESBL-PH-2018, are at a competitive disadvantage.
Using the strict thresholds set, we identified plasmids similar to pESBL-PH-2018, detected during a nosocomial outbreak in the United Kingdom. The strict thresholds applied, however, may underestimate the true prevalence of the ESBL-encoding plasmid. For example, we recently identified an ESBL-encoding plasmid, pEBM1, which underwent co-integrate formation with another
blaCTX-M-15-encoding plasmid from a
K. pneumoniae strain [
43]. A plasmid co-integrate may not be recognized as highly similar using the similarity thresholds. Our approach was designed to detect very similar plasmids, with a similar number of AMR genes; only four plasmids detected were missing four or fewer AMR genes from the 11 present on pESBL-PH-2018. However, despite this, the strict thresholds applied, definitively prove that plasmids very closely related to pESBL-PH-2018 have a wide host range, including across bacterial genera and species and display remarkable conservation over a 10-year period, existing in diverse strains with an extensive drug-resistant profile.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}