
Manipulating the Microbiome: An Alternative Treatment for Bile Acid Diarrhoea

 , , , , and
, , , , and 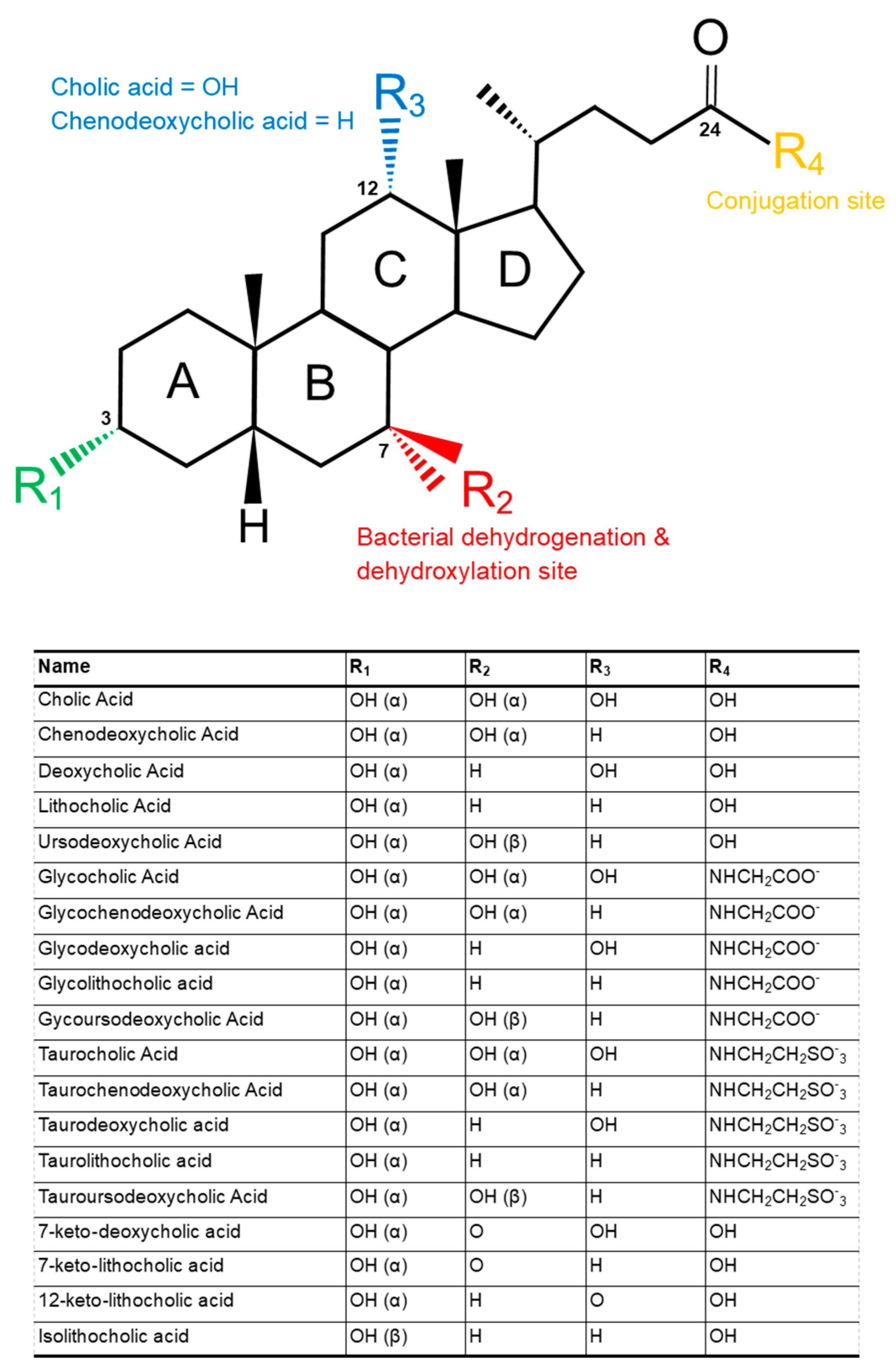{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- Type I, when the ileum is damaged due to inflammation or surgical removal.
- Type II is idiopathic and is also known as “primary BAD”.
- Type III, results from another disease or condition (such as gallbladder removal).
2. Structure and Function of Bile Acids
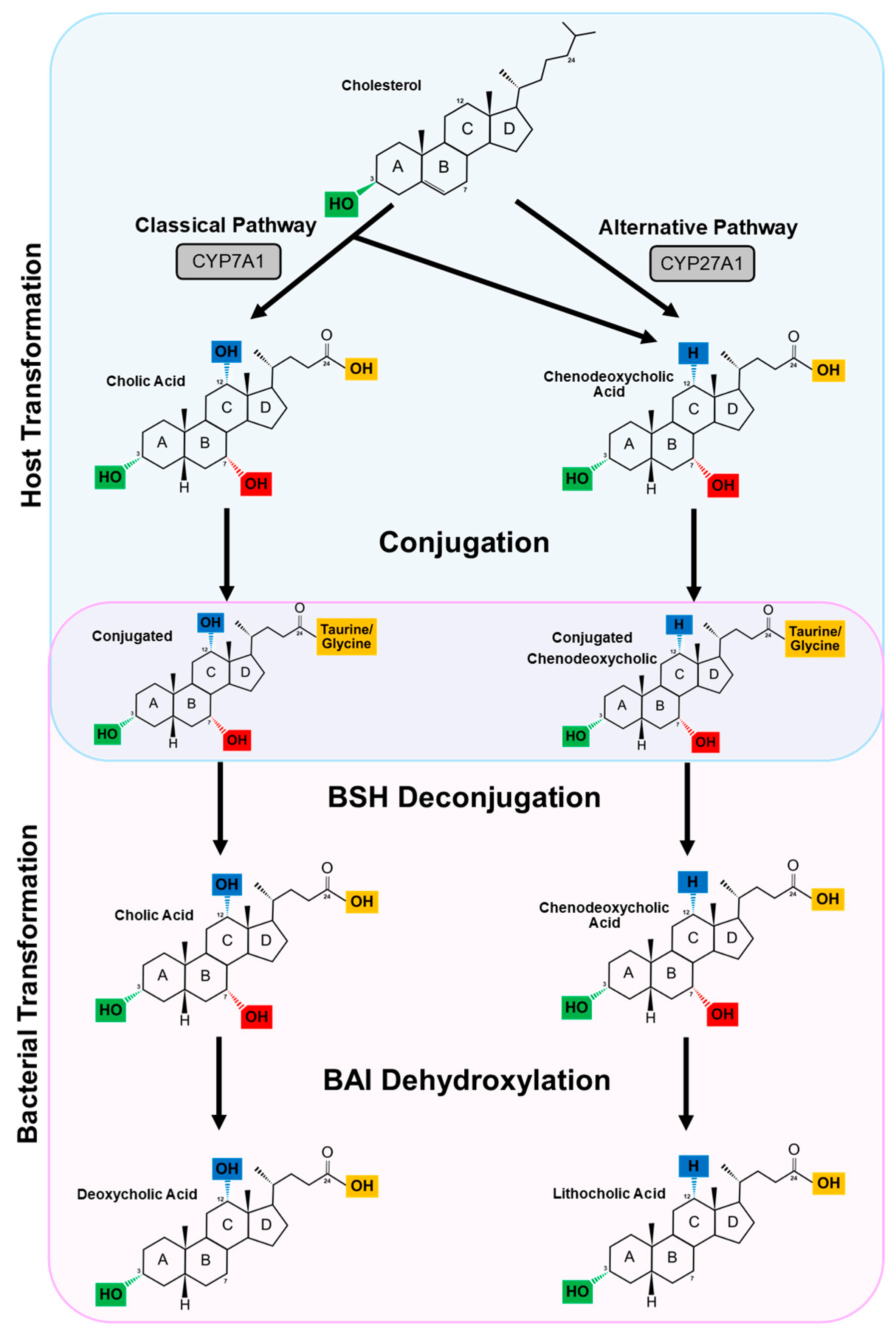
3. The Enterohepatic Circulation and Primary Bile Acid Synthesis
4. Secondary Bile Acids
4.1. Deconjugation
4.2. Dehydroxylation and Dehydrogenation
4.2.1. Dehydroxylation
4.2.2. Dehydrogenation
4.3. H+ Bile Acid Transporter
5. Bile Acid Receptors and Downstream Targets
5.1. Bile Acid Regulate Their Own Synthesis
5.2. Farnesoid X Receptor
5.3. Diet1 Protein
5.4. TGR5 Receptor
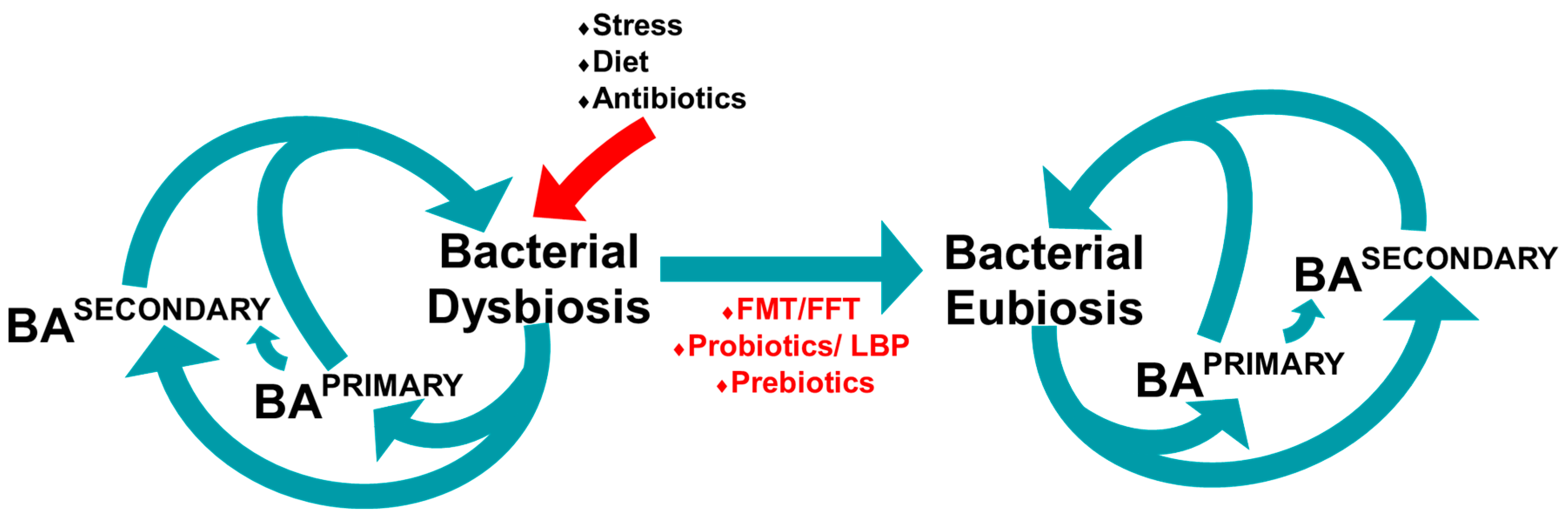
6. Microbiome
6.1. Gut Microbiome
6.2. Microbiome and Bile Acid Profile in Mice
6.3. The Gut Microbiome and Bile Acid Diarrhoea
7. Manipulating the Microbiome
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arasaradnam, R.P.; Brown, S.; Forbes, A.; Fox, M.R.; Hungin, P.; Kelman, L.; Major, G.; O’Connor, M.; Sanders, D.S.; Sinha, R.; et al. Guidelines for the investigation of chronic diarrhoea in adults: British Society of Gastroenterology, 3rd edition. Gut 2018, 67, 1380–1399. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.R.F.; Arasaradnam, R.; Andreyev, H.J.N. Diagnosis and management of bile acid diarrhoea: A survey of UK expert opinion and practice. Front. Gastroenterol. 2020, 11, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Bannaga, A.; Kelman, L.; O’Connor, M.; Pitchford, C.; Walters, J.R.F.; Arasaradnam, R.P. How bad is bile acid diarrhoea: An online survey of patient-reported symptoms and outcomes. BMJ Open Gastroenterol. 2017, 4, e000116. [Google Scholar] [CrossRef]
- Wedlake, L.; A’Hern, R.; Russell, D.; Thomas, K.; Walters, J.R.F.; Andreyev, H.J.N. Systematic review: The prevalence of idiopathic bile acid malabsorption as diagnosed by SeHCAT scanning in patients with diarrhoea-predominant irritable bowel syndrome. Aliment. Pharmacol. Ther. 2009, 30, 707–717. [Google Scholar] [CrossRef]
- Slattery, S.A.; Niaz, O.; Aziz, Q.; Ford, A.C.; Farmer, A.D. Systematic review with meta-analysis: The prevalence of bile acid malabsorption in the irritable bowel syndrome with diarrhoea. Aliment. Pharmacol. Ther. 2015, 42, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Cherian, P.; Raju, G.S.; Dawson, B.F.; Mahon, S.; Bardhan, K.D. Bile acid malabsorption in persistent diarrhoea. J. R. Coll. Physicians Lond. 2000, 34, 448–451. [Google Scholar] [PubMed]
- Williams, A.J.; Merrick, M.V.; Eastwood, M.A. Idiopathic bile acid malabsorption: A review of clinical presentation, diagnosis, and response to treatment. Gut 1991, 32, 1004–1006. [Google Scholar] [CrossRef]
- Walters, J.R.; Tasleem, A.M.; Omer, O.S.; Brydon, W.G.; Dew, T.; le Roux, C.W. A New Mechanism for Bile Acid Diarrhea: Defective Feedback Inhibition of Bile Acid Biosynthesis. Clin. Gastroenterol. Hepatol. 2009, 7, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Poley, J.R. Role of Bile Acid Malabsorption in Pathogenesis of Diarrhea and Steatorrhea in Patients with Ileal Resection. Gastroenterology 1972, 62, 918–934. [Google Scholar] [CrossRef]
- Aldini, R.; Roda, A.; Festi, D.; Sama, C.; Mazzella, G.; Bazzoli, F.; Morselli, A.M.; Roda, E.; Barbara, L. Bile acid malabsorption and bile acid diarrhea in intestinal resection. Dig. Dis. Sci. 1982, 27, 495–502. [Google Scholar] [CrossRef]
- Joyce, S.A.; Gahan, C.G. Disease-Associated Changes in Bile Acid Profiles and Links to Altered Gut Microbiota. Dig. Dis. 2017, 35, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Riemsma, R.; Al, M.; Ramos, I.C.; Deshpande, S.N.; Armstrong, N.; Lee, Y.C.; Ryder, S.; Noake, C.; Krol, M.; Oppe, M.; et al. SeHCAT [tauroselcholic (selenium-75) acid] for the investigation of bile acid malabsorption and measurement of bile acid pool loss: A systematic review and cost-effectiveness analysis. Heal. Technol. Assess. 2013, 17, 1–236. [Google Scholar] [CrossRef]
- Vaiopoulou, A. Molecular basis of the irritable bowel syndrome. World J. Gastroenterol. 2014, 20, 376–383. [Google Scholar] [CrossRef]
- Bosaeus, I.; Andersson, H.; Nyström, C. Effect of a Low-Fat Diet on Bile Salt Excretion and Diarrhoea in the Gastrointestinal Radiation Syndrome. Acta Radiol. Oncol. Radiat. Phys. Biol. 1979, 18, 460–464. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Chang, E.B. Interactions between Diet, Bile Acid Metabolism, Gut Microbiota, and Inflammatory Bowel Diseases. Dig. Dis. 2015, 33, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Poley, J.R. Cholestyramine Treatment of Diarrhea Associated with Ileal Resection. N. Engl. J. Med. 1969, 281, 397–402. [Google Scholar] [CrossRef]
- Rössel, P.; Sortsøe Jensen, H.; Qvist, P.; Arveschoug, A. Prognosis of Adult-Onset Idiopathic Bile Acid Malabsorption. Scand. J. Gastroenterol. 1999, 34, 587–590. [Google Scholar] [CrossRef]
- Farina, A.; Dumonceau, J.M.; Lescuyer, P. Proteomic analysis of human bile and potential applications for cancer diagnosis. Expert Rev. Proteom. 2009, 6, 285–301. [Google Scholar] [CrossRef]
- Falany, C.N.; Johnson, M.R.; Barnes, S.; Diasio, R.B. Glycine and taurine conjugation of bile acids by a single enzyme. Molecular cloning and expression of human liver bile acid CoA:amino acid N-acyltransferase. J. Biol. Chem. 1994, 269, 205–934. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Stieger, B.; Hagenbuch, B.; Meier, P.J. Hepatic Transport of Bile Salts. Semin. Liver Dis. 2000, 20, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A. The function of bile salts in fat absorption. Biochem. J. 1963, 89, 57–68. [Google Scholar] [CrossRef]
- Houten, S.M.; Watanabe, M.; Auwerx, J. Endocrine functions of bile acids. EMBO J. 2006, 25, 1419–1425. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of Bile Acids and Bile Acid Receptors in Metabolic Regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Lowe, P.J.; Billington, D. Membrane lipid composition and susceptibility to bile salt damage. Biochim. Biophys. Acta Biomembr. 1980, 599, 294–300. [Google Scholar] [CrossRef]
- Zavaglia, A.G.; Kociubinski, G.; Perez, P.; Disalvo, E.; De Antoni, G. Effect of bile on the lipid composition and surface properties of bifidobacteria. J. Appl. Microbiol. 2002, 93, 794–799. [Google Scholar] [CrossRef]
- Sagar, N.M.; Duboc, H.; Kay, G.L.; Tauqeer, A.; Wicaksono, A.N.; Covington, J.A.; Quince, C.; Kokkorou, M.; Svolos, V.; Palmieri, L.J.; et al. The Pathophysiology of Bile Acid Diarrhoea: Differences in the Colonic Microbiome, Metabolome and Bile Acids. SSRN Electron. J. 2020, 10. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, T.; Zhao, A.; Wang, X.; Xie, G.; Huang, F.; Liu, J.; Zhao, Q.; Wang, S.; Wang, C.; et al. The Brain Metabolome of Male Rats across the Lifespan. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Hofmann, A.F. The Continuing Importance of Bile Acids in Liver and Intestinal Disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef]
- Vlahcevic, Z.R.; Heuman, D.M.; Hylemon, P.B. Physiology and pathophysiology of enterohepatic circulation of bile acids. In Hepatology: A Textbook of Liver Disease, 3rd ed.; Zakim, D., Boyer, T., Eds.; Saunders: Philadelphia, PA, USA, 1996. [Google Scholar]
- Russell, D.W. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Reihnér, E.; Björkhem, I.; Angelin, B.; Ewerth, S.; Einarsson, K. Bile acid synthesis in humans: Regulation of hepatic microsomal cholesterol 7α-hydroxylase activity. Gastroenterology 1989, 97, 1498–1505. [Google Scholar] [CrossRef]
- Schwarz, M.; Russell, D.W.; Dietschy, J.M.; Turley, S.D. Marked reduction in bile acid synthesis in cholesterol 7α-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J. Lipid Res. 1998, 39, 1833–1843. [Google Scholar] [CrossRef]
- Gälman, C.; Arvidsson, I.; Angelin, B.; Rudling, M. Monitoring hepatic cholesterol 7α-hydroxylase activity by assay of the stable bile acid intermediate 7α-hydroxy-4-cholesten-3-one in peripheral blood. J. Lipid Res. 2003, 44, 859–866. [Google Scholar] [CrossRef]
- Axelson, M.; Aly, A.; Sjövall, J. Levels of 7α-hydroxy-4-cholesten-3-one in plasma reflect rates of bile acid synthesis in man. FEBS Lett. 1988, 239, 324–328. [Google Scholar] [CrossRef]
- Camilleri, M.; Nadeau, A.; Tremaine, W.J.; Lamsam, J.; Burton, D.; Odunsi, S.; Sweetser, S.; Singh, R. Measurement of serum 7α-hydroxy-4-cholesten-3-one (or 7αC4), a surrogate test for bile acid malabsorption in health, ileal disease and irritable bowel syndrome using liquid chromatography-tandem mass spectrometry. Neurogastroenterol. Motil. 2009, 21, 734-e43. [Google Scholar] [CrossRef] [PubMed]
- Batta, A.K.; Salen, G.; Shefer, S. Substrate specificity of cholylglycine hydrolase for the hydrolysis of bile acid conjugates. J. Biol. Chem. 1984, 259, 15036–15039. [Google Scholar] [CrossRef]
- Kawamoto, K.; Horibe, I.; Uchida, K. Purification and Characterization of a New Hydrolase for Conjugated Bile Acids, Chenodeoxycholyltaurine Hydrolase, from Bacteroides vulgatus. J. Biochem. 1989, 106, 1049–1053. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Corzo, G.; Gilliland, S. Bile Salt Hydrolase Activity of Three Strains of Lactobacillus acidophilus. J. Dairy Sci. 1999, 82, 472–480. [Google Scholar] [CrossRef]
- Tanaka, H.; Hashiba, H.; Kok, J.; Mierau, I. Bile Salt Hydrolase of Bifidobacterium longum—Biochemical and Genetic Characterization. Appl. Environ. Microbiol. 2000, 66, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Franz, C.M.; Specht, I.; Haberer, P.; Holzapfel, W.H. Bile Salt Hydrolase Activity of Enterococci Isolated from Food: Screening and Quantitative Determination. J. Food Prot. 2001, 64, 725–729. [Google Scholar] [CrossRef]
- Masuda, N. Deconjugation of Bile Salts by Bacteroides and Clostridium. Microbiol. Immunol. 1981, 25, 1–11. [Google Scholar] [CrossRef]
- Kishinaka, M.; Umeda, A.; Kuroki, S. High concentrations of conjugated bile acids inhibit bacterial growth of Clostridium perfringens and induce its extracellular cholyglycine hydrolase. Steroids 1994, 59, 485–489. [Google Scholar] [CrossRef]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 2019, 7, 1–16. [Google Scholar] [CrossRef]
- Tian, Y.; Cai, J.; Gui, W.; Nichols, R.G.; Koo, I.; Zhang, J.; Anitha, M.; Patterson, A.D. Berberine Directly Affects the Gut Microbiota to Promote Intestinal Farnesoid X Receptor Activation. Drug Metab. Dispos. 2019, 47, 86–93. [Google Scholar] [CrossRef]
- Begley, M.; Hill, C.; Gahan, C.G.M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef]
- De Smet, I.; Van Hoorde, L.; Woestyne, M.V.; Christiaens, H.; Verstraete, W. Significance of bile salt hydrolytic activities of lactobacilli. J. Appl. Bacteriol. 1995, 79, 292–301. [Google Scholar] [CrossRef]
- Dussurget, O.; Cabanes, D.; Dehoux, P.; Lecuit, M.; Buchrieser, C.; Glaser, P.; Cossart, P. The European Listeria Genome Consortium Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol. Microbiol. 2002, 45, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Sannasiddappa, T.H.; Lund, P.A.; Clarke, S.R.; Sannasiddappa, T. In Vitro Antibacterial Activity of Unconjugated and Conjugated Bile Salts on Staphylococcus aureus. Front. Microbiol. 2017, 8, 1581. [Google Scholar] [CrossRef]
- Tian, Y.; Gui, W.; Koo, I.; Smith, P.B.; Allman, E.L.; Nichols, R.G.; Rimal, B.; Cai, J.; Liu, Q.; Patterson, A.D. The microbiome modulating activity of bile acids. Gut Microbes 2020, 11, 979–996. [Google Scholar] [CrossRef]
- Masuda, N.; Oda, H.; Hirano, S.; Masuda, M.; Tanaka, H. 7 α-Dehydroxylation of bile acids by resting cells of a Eubacterium lentum-like intestinal anaerobe, strain c-25. Appl. Environ. Microbiol. 1984, 47, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.E.; Berr, F.; A Thomas, L.; Dowling, R.; Hylemon, P.B. Isolation and characterization of cholic acid 7α-dehydroxylating fecal bacteria from cholesterol gallstone patients. J. Hepatol. 2000, 32, 4–10. [Google Scholar] [CrossRef]
- Björkhem, I.; Einarsson, K.; Melone, P.; Hylemon, P. Mechanism of intestinal formation of deoxycholic acid from cholic acid in humans: Evidence for a 3-oxo-delta 4-steroid intermediate. J. Lipid Res. 1989, 30, 1033–1039. [Google Scholar] [CrossRef]
- Vital, M.; Rud, T.; Rath, S.; Pieper, D.H.; Schlüter, D. Diversity of Bacteria Exhibiting Bile Acid-inducible 7α-dehydroxylation Genes in the Human Gut. Comput. Struct. Biotechnol. J. 2019, 17, 1016–1019. [Google Scholar] [CrossRef]
- Stellwag, E.J.; Hylemon, P.B. 7α Dehydroxylation of cholic acid and chenodeoxycholic acid by Clostridium leptum. J. Lipid Res. 1979, 20, 325–333. [Google Scholar] [CrossRef]
- Batta, A.K.; Salen, G.; Arora, R.; Shefer, S.; Batta, M.; Person, A. Side chain conjugation prevents bacterial 7-dehydroxylation of bile acids. J. Biol. Chem. 1990, 265, 10925–10928. [Google Scholar] [CrossRef]
- Sutherland, J.D.; Macdonald, I.A. The metabolism of primary, 7-oxo, and 7β-hydroxy bile acids by Clostridium absonum. J. Lipid Res. 1982, 23, 726–732. [Google Scholar] [CrossRef]
- Macdonald, I.A.; Hutchison, D.M.; Forrest, T.P. Formation of urso-and ursodeoxy-cholic acids from primary bile acids by Clostridium absonum. J. Lipid Res. 1981, 22, 458–466. [Google Scholar] [CrossRef]
- Macdonald, I.A.; Roach, P.D. Bile salt induction of 7α-and 7β-hydroxysteroid dehydrogenases in Clostridium absonum. Biochim. Biophys. Acta Lipids Lipid Metab. 1981, 665, 262–269. [Google Scholar] [CrossRef]
- Hirano, S.; Masuda, N. Epimerization of the 7-hydroxy group of bile acids by the combination of two kinds of microorganisms with 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activity, respectively. J. Lipid Res. 1981, 22, 1060–1068. [Google Scholar] [CrossRef]
- Sherrod, J.; Hylemon, P. Partial purification and characterization of NAD-dependent 7α-hydroxysteroid dehydrogenase from Bacteroides thetaiotaomicron. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1977, 486, 351–358. [Google Scholar] [CrossRef]
- Macdonald, I.A.; Meier, E.C.; Mahony, D.E.; Costain, G.A. 3α-, 7α- And 12α-hydroxysteroid dehydrogenase activities from Clostridium perfringens. Biochim. Biophys. Acta Lipids Lipid Metab. 1976, 450, 142–153. [Google Scholar] [CrossRef]
- Bhowmik, S.; Jones, D.H.; Chiu, H.; Park, I.; Chiu, H.; Axelrod, H.L.; Farr, C.L.; Tien, H.J.; Agarwalla, S.; Lesley, S.A. Structural and functional characterization of BaiA, an enzyme involved in secondary bile acid synthesis in human gut microbe. Proteins Struct. Funct. Bioinform. 2014, 82, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.E.; Hylemon, P.B. Identification and Characterization of a Bile Acid 7α-Dehydroxylation Operon in Clostridium sp. Strain TO-931, a Highly Active 7α-Dehydroxylating Strain Isolated from Human Feces. Appl. Environ. Microbiol. 2000, 66, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.A.; Mahony, D.E.; Jellet, J.F.; Meier, C. Nad-dependent 3α- and 12α-hydroxysteroid dehydrogenase activities from eubacterwm lentum atcc no. 25559. Biochim. Biophys. Acta Lipids Lipid Metab. 1977, 489, 466–476. [Google Scholar] [CrossRef]
- Edenharder, R.; Pfützner, A.; Hammann, R. Characterization of NAD-dependent 3α- and 3β-hydroxysteroid dehydrogenase and of NADP-dependent 7β-hydroxysteroid dehydrogenase from Peptostreptococcus productus. Biochim. Biophys. Acta Lipids Lipid Metab. 1989, 1004, 230–238. [Google Scholar] [CrossRef]
- Edenharder, R.; Pfützner, M.; Hammann, R. NADP-dependent 3β-, 7α- and 7β-hydroxysteroid dehydrogenase activities from a lecithinase-lipase-negative Clostridium species 25.11.c. Biochim. Biophys. Acta Lipids Lipid Metab. 1989, 1002, 37–44. [Google Scholar] [CrossRef]
- Edenharder, R.; Pfützner, M. Partial purification and characterization of an NAD-dependent 3 beta-hydroxysteroid dehydrogenase from Clostridium innocuum. Appl. Environ. Microbiol. 1989, 55, 1656. [Google Scholar] [CrossRef]
- Teruaki, A.; Taiko, A.; Masao, H.; Tsuneo, N.; Kyoichi, K. Enzymes involved in the formation of 3β,7β-dihydroxy-12-oxo-5β-cholanic acid from dehydrocholic acid by Ruminococcus sp. obtained from human intestine. Biochim. Biophys. Acta Lipids Lipid Metab. 1987, 921, 275–280. [Google Scholar] [CrossRef]
- Sutherland, J.D.; Williams, C.N. Bile acid induction of 7 alpha- and 7 beta-hydroxysteroid dehydrogenases in Clostridium limosum. J. Lipid Res. 1985, 26, 344–350. [Google Scholar] [CrossRef]
- Stellwag, E.; Hylemon, P. Purification and characterization of bile salt hydrolase from Bacteroides fragilis subsp. fragilis. Biochim. Biophys. Acta Enzym. 1976, 452, 165–176. [Google Scholar] [CrossRef]
- Coleman, J.P.; Hudson, L.L.; Adams, M.J. Characterization and regulation of the NADP-linked 7 alpha-hydroxysteroid dehydrogenase gene from Clostridium sordellii. J. Bacteriol. 1994, 176, 4865–4874. [Google Scholar] [CrossRef]
- Hirano, S.; Masuda, N.; Oda, H.; Mukai, H. Transformation of bile acids by Clostridium perfringens. Appl. Environ. Microbiol. 1981, 42, 394–399. [Google Scholar] [CrossRef]
- Heuman, D.M. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J. Lipid Res. 1989, 30, 719–730. [Google Scholar] [CrossRef]
- Heuman, U.M.; Pandak, W.M.; Hylemon, P.B.; Vlahcevic, Z.R. Conjugates of ursodeoxycholate protect against cytotoxicity of more hydrophobic bile salts: In vitro studies in rat hepatocytes and human erythrocytes. Hepatology 1991, 14, 920–926. [Google Scholar] [CrossRef]
- Harris, J.; Hylemon, P. Partial purification and characterization of NADP-dependent 12α-hydroxysteroid dehydrogenase from clostridium leptum. Biochim. Biophys. Acta Lipids Lipid Metab. 1978, 528, 148–157. [Google Scholar] [CrossRef]
- Macdonald, I.A.; Jellett, J.F.; Mahony, D.E. 12alpha-Hydroxysteroid dehydrogenase from Clostridium group P strain C48-50 ATCC No. 29733: Partial purification and characterization. J. Lipid Res. 1979, 20, 234–239. [Google Scholar] [CrossRef]
- Edenharder, R.; Schneider, J. 12 beta-dehydrogenation of bile acids by Clostridium paraputrificum, C. tertium, and C. difficile and epimerization at carbon-12 of deoxycholic acid by cocultivation with 12 alpha-dehydrogenating Eubacterium lentum. Appl. Environ. Microbiol. 1985, 49, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Edenharder, R.; Pfützner, A. Characterization of NADP-dependent 12β-hydroxysteroid dehydrogenase from Clostridium paraputrificum. Biochim. Biophys. Acta Lipids Lipid Metab. 1988, 962, 362–370. [Google Scholar] [CrossRef]
- Mallonee, D.H.; Hylemon, P.B. Sequencing and expression of a gene encoding a bile acid transporter from Eubacterium sp. strain VPI 12708. J. Bacteriol. 1996, 178, 7053–7058. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nat. Cell Biol. 2020, 582, 566–570. [Google Scholar] [CrossRef]
- Distrutti, E.; Santucci, L.; Cipriani, S.; Renga, B.; Schiaroli, E.; Ricci, P.; Donini, A.; Fiorucci, S. Bile acid activated receptors are targets for regulation of integrity of gastrointestinal mucosa. J. Gastroenterol. 2015, 50, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Seol, W.; Choi, H.S.; Moore, D.D. Isolation of proteins that interact specifically with the retinoid X receptor: Two novel orphan receptors. Mol. Endocrinol. 1995, 9, 72–85. [Google Scholar] [CrossRef]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous Bile Acids Are Ligands for the Nuclear Receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Mueller, M.; Thorell, A.; Claudel, T.; Jha, P.; Koefeler, H.; Lackner, C.; Hoesel, B.; Fauler, G.; Stojakovic, T.; Einarsson, C.; et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J. Hepatol. 2015, 62, 1398–1404. [Google Scholar] [CrossRef]
- Downes, M.; Verdecia, M.A.; Roecker, A.; Hughes, R.; HogenEsch, J.B.; Kast-Woelbern, H.R.; Bowman, M.E.; Ferrer, J.L.; Anisfeld, A.M.; Edwards, P.A.; et al. A Chemical, Genetic, and Structural Analysis of the Nuclear Bile Acid Receptor FXR. Mol. Cell 2003, 11, 1079–1092. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, W.; Chen, Y.; Huang, F.; Lu, L.; Lin, C.; Huang, T.; Ning, Z.; Zhai, L.; Zhong, L.L.; et al. A Clostridia-rich microbiota enhances bile acid excretion in diarrhea-predominant irritable bowel syndrome. J. Clin. Investig. 2019, 130, 438–450. [Google Scholar] [CrossRef]
- Trefflich, I.; Marschall, H.-U.; di Giuseppe, R.; Ståhlman, M.; Michalsen, A.; Lampen, A.; Abraham, K.; Weikert, C. Associations between Dietary Patterns and Bile Acids: Results from a Cross-Sectional Study in Vegans and Omnivores. Nutrients 2019, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-beta-muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Evanst, R.M. The RXR Heterodimers and Orphan Receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Dawson, M.I.; Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 21–56. [Google Scholar] [CrossRef]
- Kong, B.; Wang, L.; Chiang, J.Y.L.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012, 56, 1034–1043. [Google Scholar] [CrossRef]
- Zollner, G.; Marschall, H.U.; Wagner, A.M.; Trauner, M. Role of Nuclear Receptors in the Adaptive Response to Bile Acids and Cholestasis: Pathogenetic and Therapeutic Considerations. Mol. Pharm. 2006, 3, 231–251. [Google Scholar] [CrossRef]
- Ballatori, N.; Christian, W.V.; Lee, J.Y.; Dawson, P.A.; Soroka, C.J.; Boyer, J.L.; Madejczyk, M.S.; Li, N. OSTα-OSTβ: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005, 42, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, L.; Lee, J.M.; Chin, R.G.; Auwerx, J.; Reue, K. Diet1 Functions in the FGF15/19 Enterohepatic Signaling Axis to Modulate Bile Acid and Lipid Levels. Cell Metab. 2013, 17, 916–928. [Google Scholar] [CrossRef]
- Keitel, V.; Cupisti, K.; Ullmer, C.; Knoefel, W.T.; Kubitz, R.; Häussinger, D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 2009, 50, 861–870. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G Protein-coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Poole, D.P.; Godfrey, C.; Cattaruzza, F.; Cottrell, G.S.; Kirkland, J.G.; Pelayo, J.C.; Bunnett, N.W.; Corvera, C.U. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol. Motil. 2010, 22, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Alemi, F.; Poole, D.P.; Chiu, J.; Schoonjans, K.; Cattaruzza, F.; Grider, J.R.; Bunnett, N.W.; Corvera, C.U. The Receptor TGR5 Mediates the Prokinetic Actions of Intestinal Bile Acids and Is Required for Normal Defecation in Mice. Gastroenterology 2013, 144, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Burge, M.N. Fungi in Biological Control Systems; Manchester University Press: Manchester, UK, 1988. [Google Scholar]
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 1–3. [Google Scholar] [CrossRef]
- Simon, G.L.; Gorbach, S.L. Intestinal flora in health and disease. Gastroenterology 1984, 86, 174–193. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Marchesi, J.R. The Microbiomes of Things. 2017. Available online: https://microbiologysociety.org/publication/past-issues/the-microbiome/article/the-microbiomes-of-things.html (accessed on 28 May 2020).
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Savage, D.C. Microbial Ecology of the Gastrointestinal Tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef]
- Amos, G.C.A.; Logan, A.; Anwar, S.; Fritzsche, M.; Mate, R.; Bleazard, T.; Rijpkema, S. Developing standards for the microbiome field. Microbiome 2020, 8, 1–13. [Google Scholar] [CrossRef]
- Yoo, B.B.; Mazmanian, S.K. The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 2017, 46, 910–926. [Google Scholar] [CrossRef]
- Lazar, V.; Ditu, L.M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef]
- Furness, J.B. The Enteric Nervous System; Wiley-Blackwell: Hoboken, NJ, USA, 2006. [Google Scholar]
- Furness, J.B.; Callaghan, B.P.; Rivera, L.R.; Cho, H.-J. The Enteric Nervous System and Gastrointestinal Innervation: Integrated Local and Central Control. Adv. Exp. Med. Biol. 2014, 817, 39–71. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Tierney, B.T.; Yang, Z.; Luber, J.M.; Beaudin, M.; Wibowo, M.C.; Baek, C.; Mehlenbacher, E.; Patel, C.J.; Kostic, A.D. The Landscape of Genetic Content in the Gut and Oral Human Microbiome. Cell Host Microbe 2019, 26, 283–295.e8. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Selwyn, F.P.; Csanaky, I.L.; Zhang, Y.; Klaassen, C.D. Importance of Large Intestine in Regulating Bile Acids and Glucagon-Like Peptide-1 in Germ-Free Mice. Drug Metab. Dispos. 2015, 43, 1544–1556. [Google Scholar] [CrossRef]
- Zhang, L.; Xie, C.; Nichols, R.G.; Chan, S.H.J.; Jiang, C.; Hao, R.; Smith, P.B.; Cai, J.; Simons, M.N.; Hatzakis, E.; et al. Farnesoid X Receptor Signaling Shapes the Gut Microbiota and Controls Hepatic Lipid Metabolism. mSystems 2016, 1, e00070-16. [Google Scholar] [CrossRef]
- Just, S.; Mondot, S.; Ecker, J.; Wegner, K.; Rath, E.; Gau, L.; Streidl, T.; Hery-Arnaud, G.; Schmidt, S.; Lesker, T.R.; et al. The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 2018, 6, 134. [Google Scholar] [CrossRef]
- Nie, Y.F.; Hu, J.; Yan, X.H. Cross-talk between bile acids and intestinal microbiota in host metabolism and health. J. Zhejiang Univ. Sci. B 2015, 16, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Jiang, C.; Xie, C.; Lv, Y.; Li, J.; Krausz, K.W.; Shi, J.; Brocker, C.N.; Desai, D.; Amin, S.G.; Bisson, W.H.; et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 2015, 6, 10166. [Google Scholar] [CrossRef]
- Duboc, H.; Rainteau, D.; Rajca, S.; Humbert, L.; Farabos, D.; Maubert, M.; Grondin, V.; Jouet, P.; Bouhassira, D.; Seksik, P.; et al. Increase in fecal primary bile acids and dysbiosis in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol. Motil. 2012, 24, 513-e247. [Google Scholar] [CrossRef]
- Hugon, P.; Dufour, J.C.; Colson, P.; Fournier, P.-E.; Sallah, K.; Raoult, D. A comprehensive repertoire of prokaryotic species identified in human beings. Lancet Infect. Dis. 2015, 15, 1211–1219. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Stecher, B.; Maier, L.; Hardt, W.D. ’Blooming’ in the gut: How dysbiosis might contribute to pathogen evolution. Nat. Rev. Genet. 2013, 11, 277–284. [Google Scholar] [CrossRef]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell. Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef] [PubMed]
- Aas, J.; Gessert, C.E.; Bakken, J.S. RecurrentClostridium difficileColitis: Case Series Involving 18 Patients Treated with Donor Stool Administered via a Nasogastric Tube. Clin. Infect. Dis. 2003, 36, 580–585. [Google Scholar] [CrossRef]
- Lopez, J.; Grinspan, A. Fecal Microbiota Transplantation for Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2016, 12, 374–379. [Google Scholar]
- Langdon, A.; Crook, N.; Dantas, G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Garborg, K.; Waagsbø, B.; Stallemo, A.; Matre, J.; Sundøy, A. Results of faecal donor instillation therapy for recurrent Clostridium difficile-associated diarrhoea. Scand. J. Infect. Dis. 2010, 42, 857–861. [Google Scholar] [CrossRef] [PubMed]
- El-Salhy, M.; Hatlebakk, J.G.; Gilja, O.H.; Kristoffersen, A.B.; Hausken, T. Efficacy of faecal microbiota transplantation for patients with irritable bowel syndrome in a randomised, double-blind, placebo-controlled study. Gut 2019, 69, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Early Clinical Trials with Live Biotherapeutic Products: Chemistry, Manufacturing, and Control Information; Guidance for Industry; Food and Drug Administration: White Oak, MD, USA, 2016. [Google Scholar]
- Fuller, R. Probiotics in human medicine. Gut 1991, 32, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Afrc, R.F. Probiotics in man and animals. J. Appl. Bacteriol. 1989, 66, 365–378. [Google Scholar] [CrossRef]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients With Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811.e7. [Google Scholar] [CrossRef]
- Foster, J.A.; Rinaman, L.; Cryan, J.F. Stress & the gut-brain axis: Regulation by the microbiome. Neurobiol. Stress 2017, 7, 124–136. [Google Scholar] [CrossRef]
- Madison, A.; Kiecolt-Glaser, J.K. Stress, depression, diet, and the gut microbiota: Human–bacteria interactions at the core of psychoneuroimmunology and nutrition. Curr. Opin. Behav. Sci. 2019, 28, 105–110. [Google Scholar] [CrossRef]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut 2016, 65, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Hardison, W.G. Hepatic Taurine Concentration and Dietary Taurine as Regulators of Bile Acid Conjugation with Taurine. Gastroenterology 1978, 75, 71–75. [Google Scholar] [CrossRef]
- Schloissnig, S.; Arumugam, M.; Sunagawa, S.; Mitreva, M.; Tap, J.; Zhu, A.; Waller, A.S.; Mende, D.R.; Kultima, J.R.; Martin, J.; et al. Genomic variation landscape of the human gut microbiome. Nat. Cell Biol. 2013, 493, 45–50. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hillman, E.B.M.; Rijpkema, S.; Carson, D.; Arasaradnam, R.P.; Wellington, E.M.H.; Amos, G.C.A. Manipulating the Microbiome: An Alternative Treatment for Bile Acid Diarrhoea. Microbiol. Res. 2021, 12, 335-353. https://doi.org/10.3390/microbiolres12020023
Hillman EBM, Rijpkema S, Carson D, Arasaradnam RP, Wellington EMH, Amos GCA. Manipulating the Microbiome: An Alternative Treatment for Bile Acid Diarrhoea. Microbiology Research. 2021; 12(2):335-353. https://doi.org/10.3390/microbiolres12020023
Chicago/Turabian StyleHillman, Evette B. M., Sjoerd Rijpkema, Danielle Carson, Ramesh P. Arasaradnam, Elizabeth M. H. Wellington, and Gregory C. A. Amos. 2021. "Manipulating the Microbiome: An Alternative Treatment for Bile Acid Diarrhoea" Microbiology Research 12, no. 2: 335-353. https://doi.org/10.3390/microbiolres12020023
APA StyleHillman, E. B. M., Rijpkema, S., Carson, D., Arasaradnam, R. P., Wellington, E. M. H., & Amos, G. C. A. (2021). Manipulating the Microbiome: An Alternative Treatment for Bile Acid Diarrhoea. Microbiology Research, 12(2), 335-353. https://doi.org/10.3390/microbiolres12020023