
Elucidating the Relations between Gut Bacterial Composition and the Plasma and Fecal Metabolomes of Antibiotic Treated Wistar Rats


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Animals and Maintenance Conditions
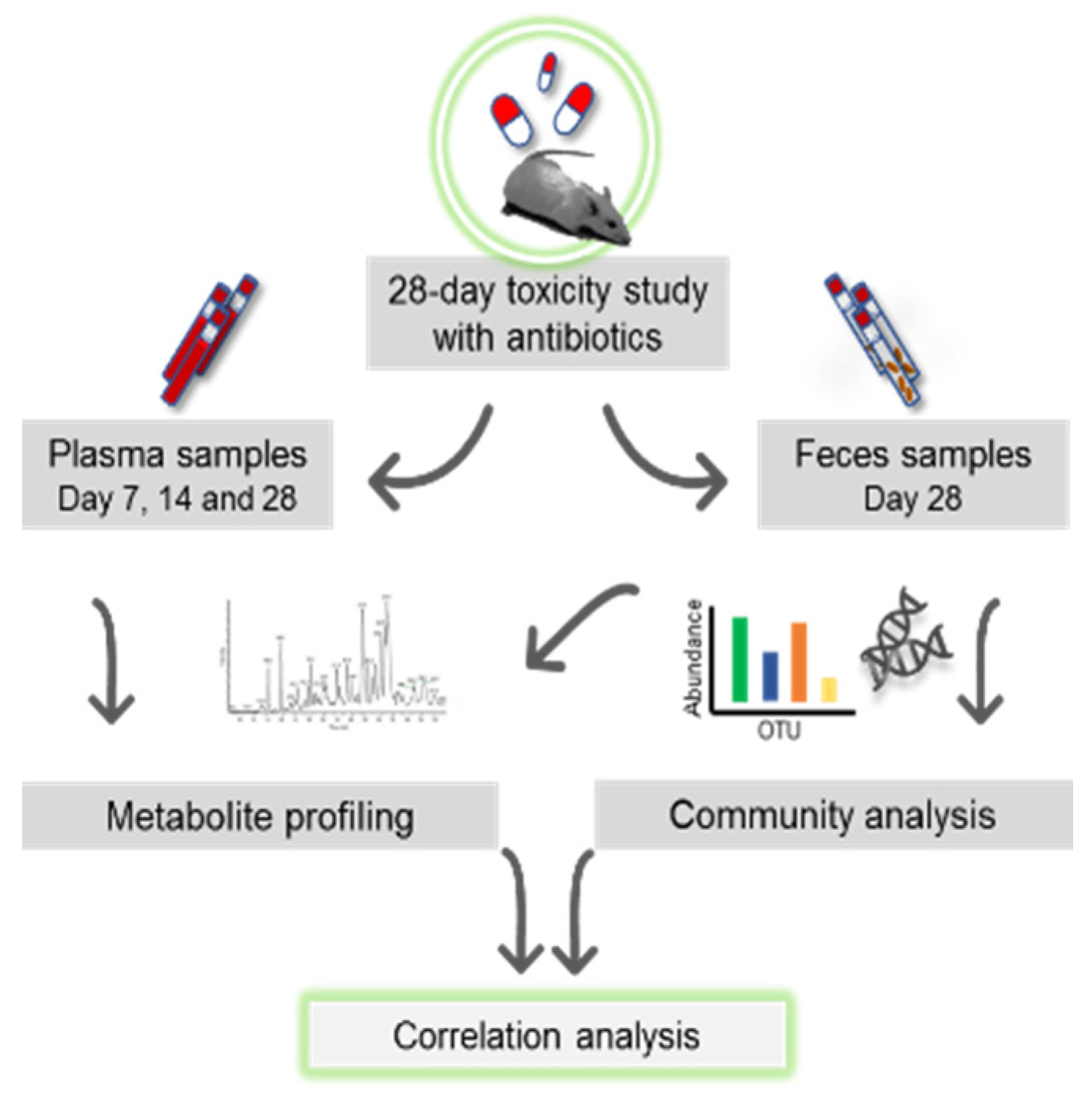
2.3. Study Design
2.4. Treatment of Animals with Antibiotics
2.5. Sampling of Plasma, Cecum and Feces for Omics Profiling
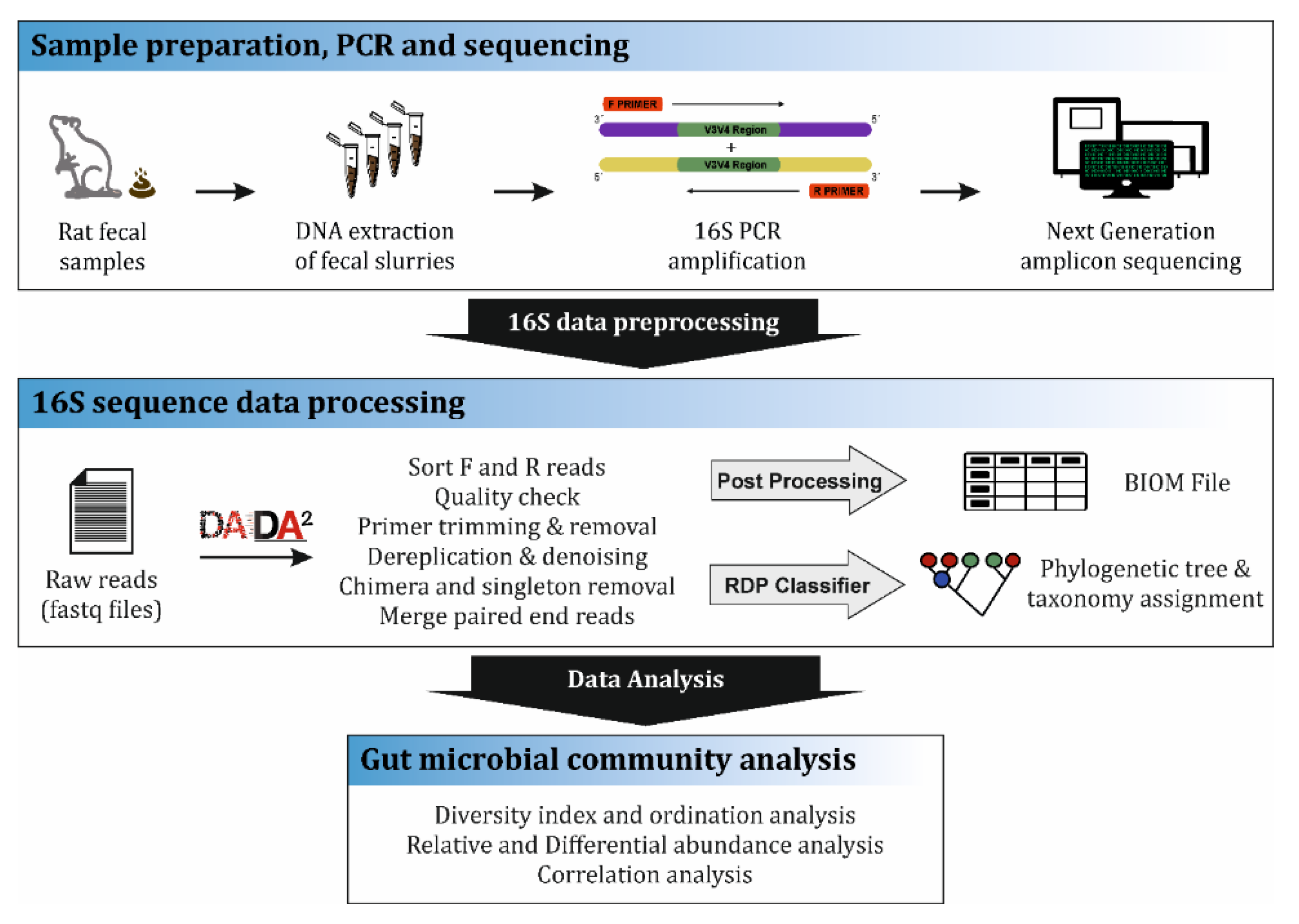
2.6. DNA Isolation and Bacterial 16S rDNA Gene Amplicon Sequencing
2.7. Metabolome Profiling of Plasma, Cecum and Fecal Matrices
2.8. Targeted Bile Acid Profiling of Plasma and Fecal Matrices
2.9. Statistics
2.10. Bioinformatics
2.11. 16S Data Normalization, Diversity and Relative Abundances Analyses
2.12. Correlation Analysis
3. Results
3.1. Clinical Signs
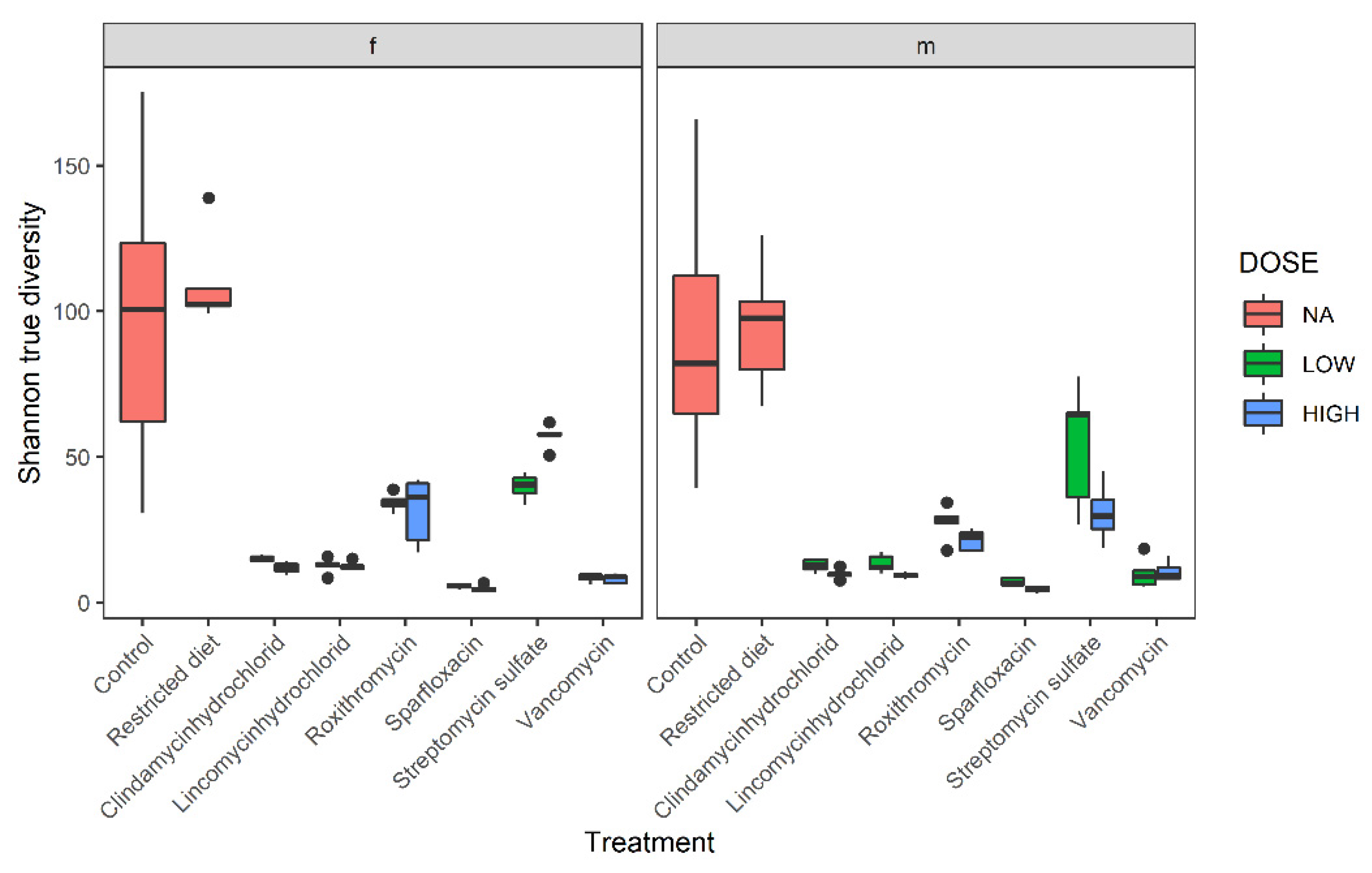
3.2. Diversity Analysis
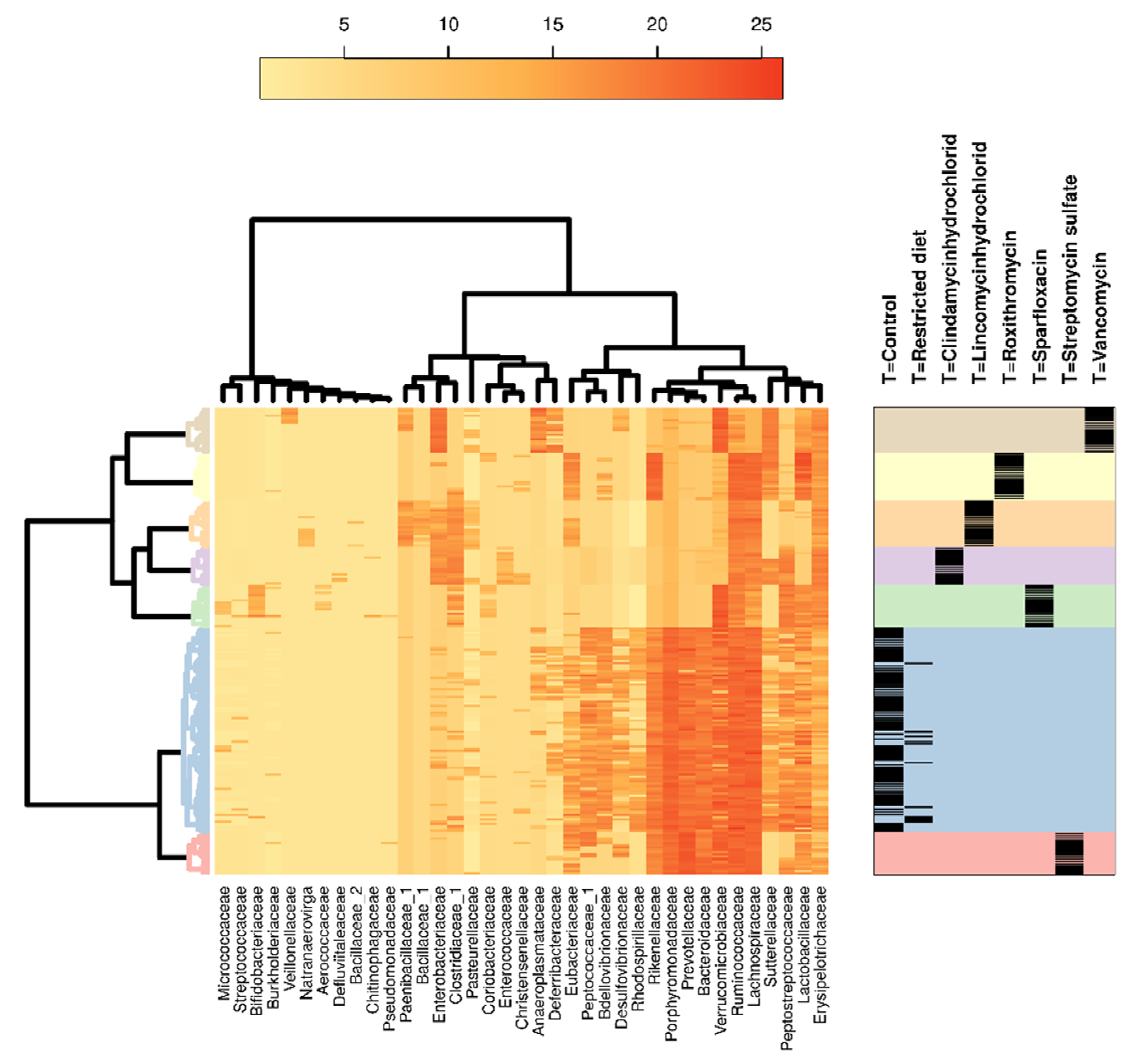
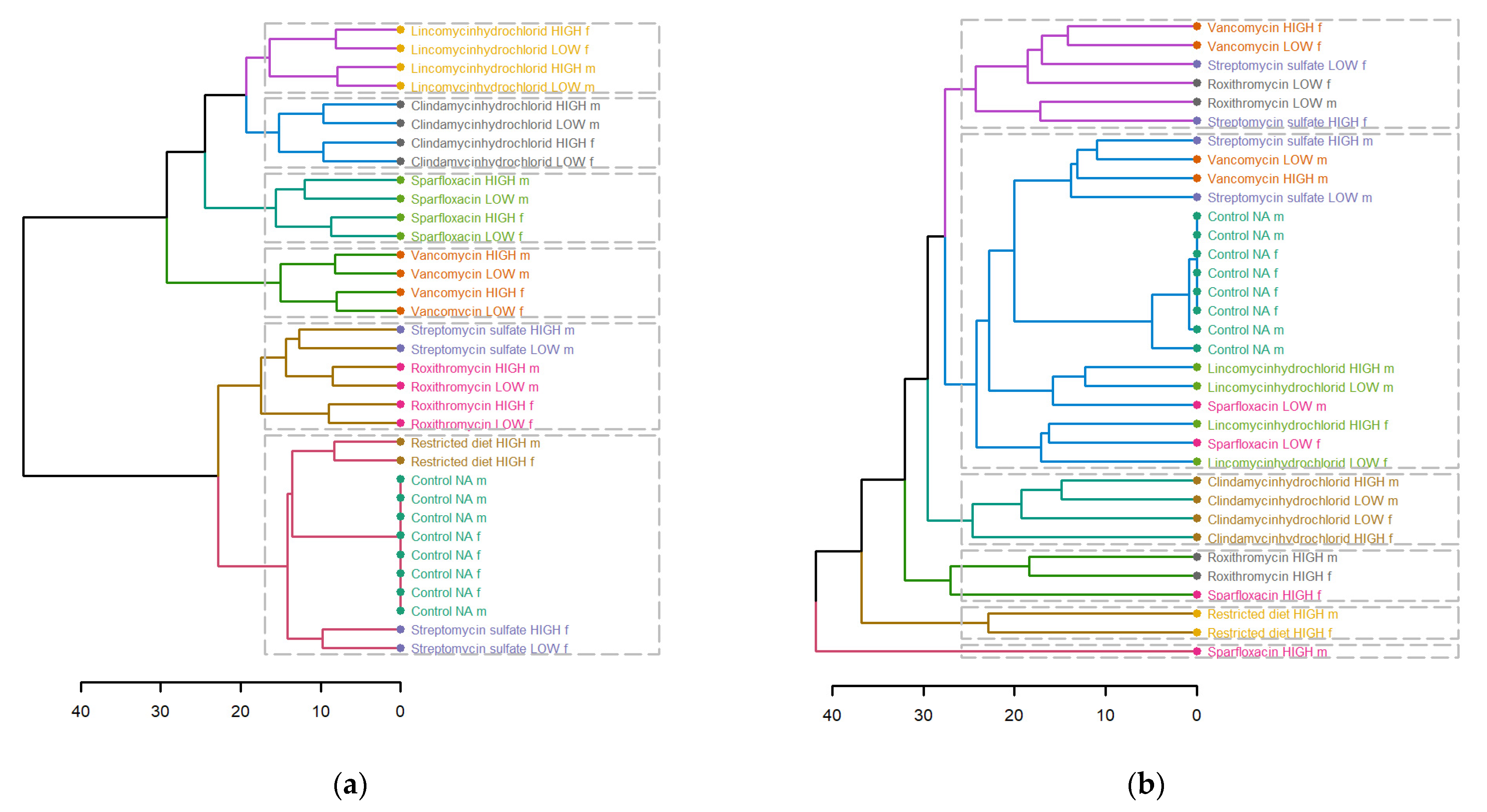
3.3. Hierarchical Clustering Analysis
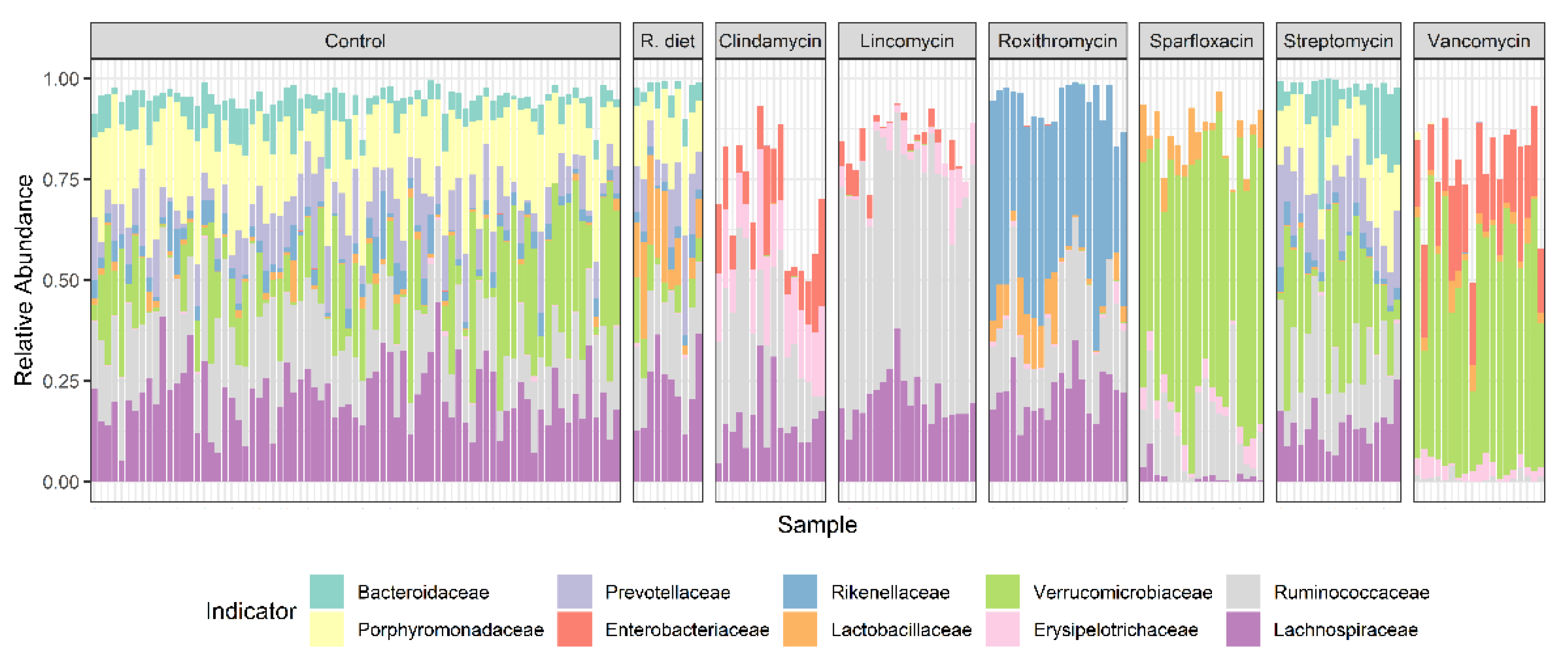
3.4. Relative Abundance Analysis
3.5. Differential Abundance Analysis
3.6. Metabolome Data Analysis
3.6.1. Fecal Metabolome
3.6.2. Plasma Metabolome
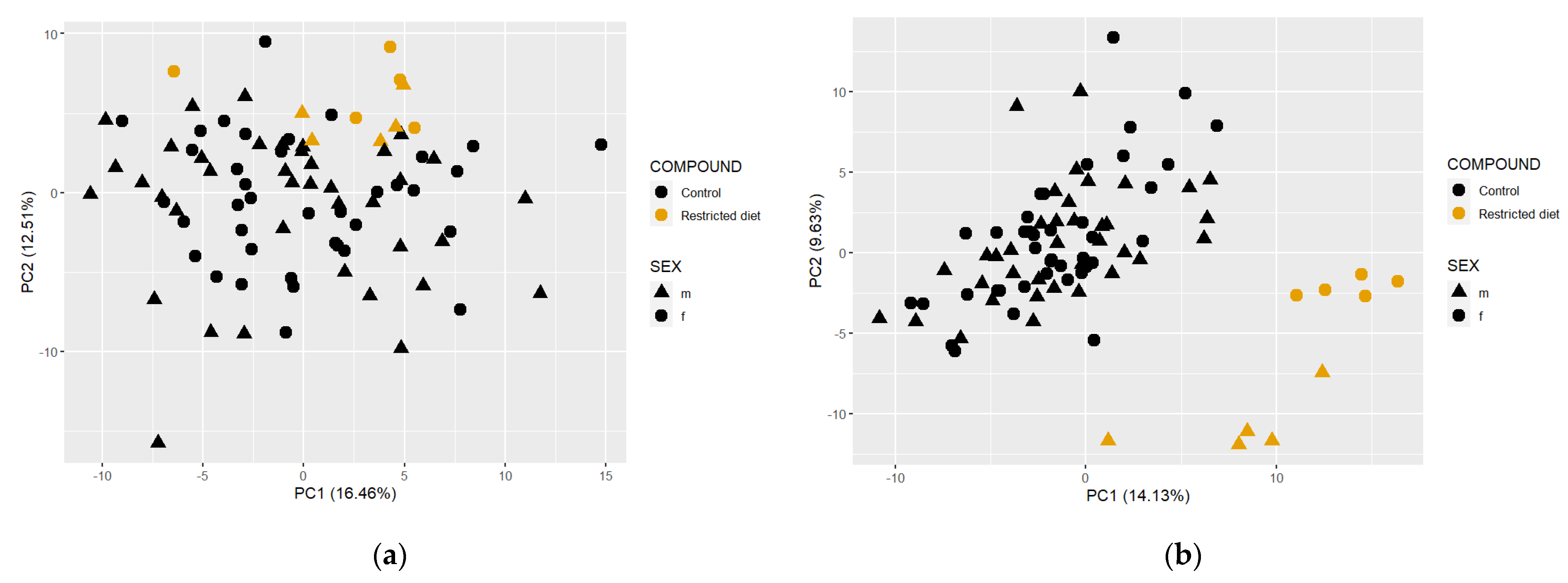
3.6.3. Controls vs. Restricted Diet in Plasma and Fecal Matrices
3.6.4. Comparison between Plasma, Feces and Cecum Metabolome Profiles
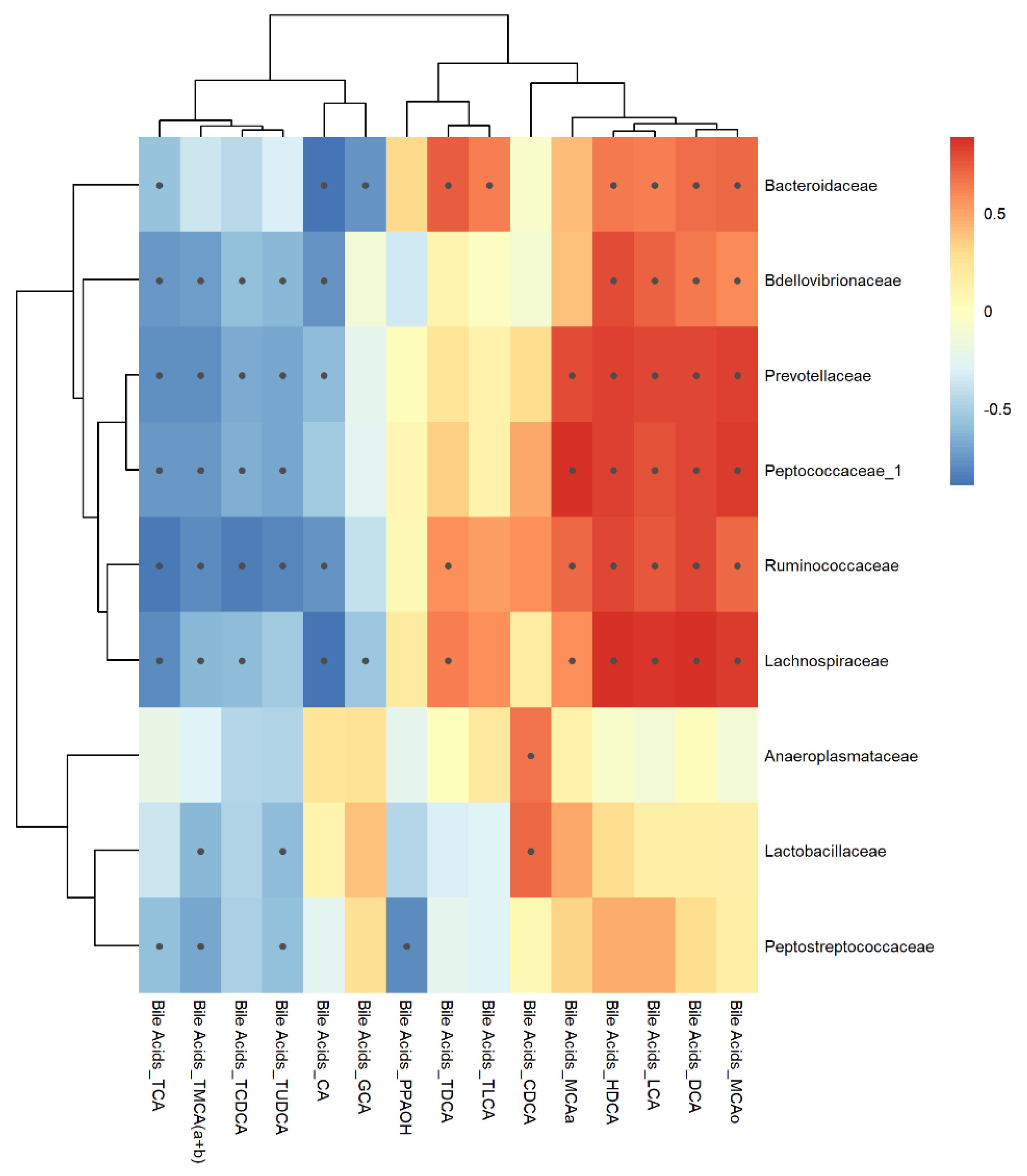
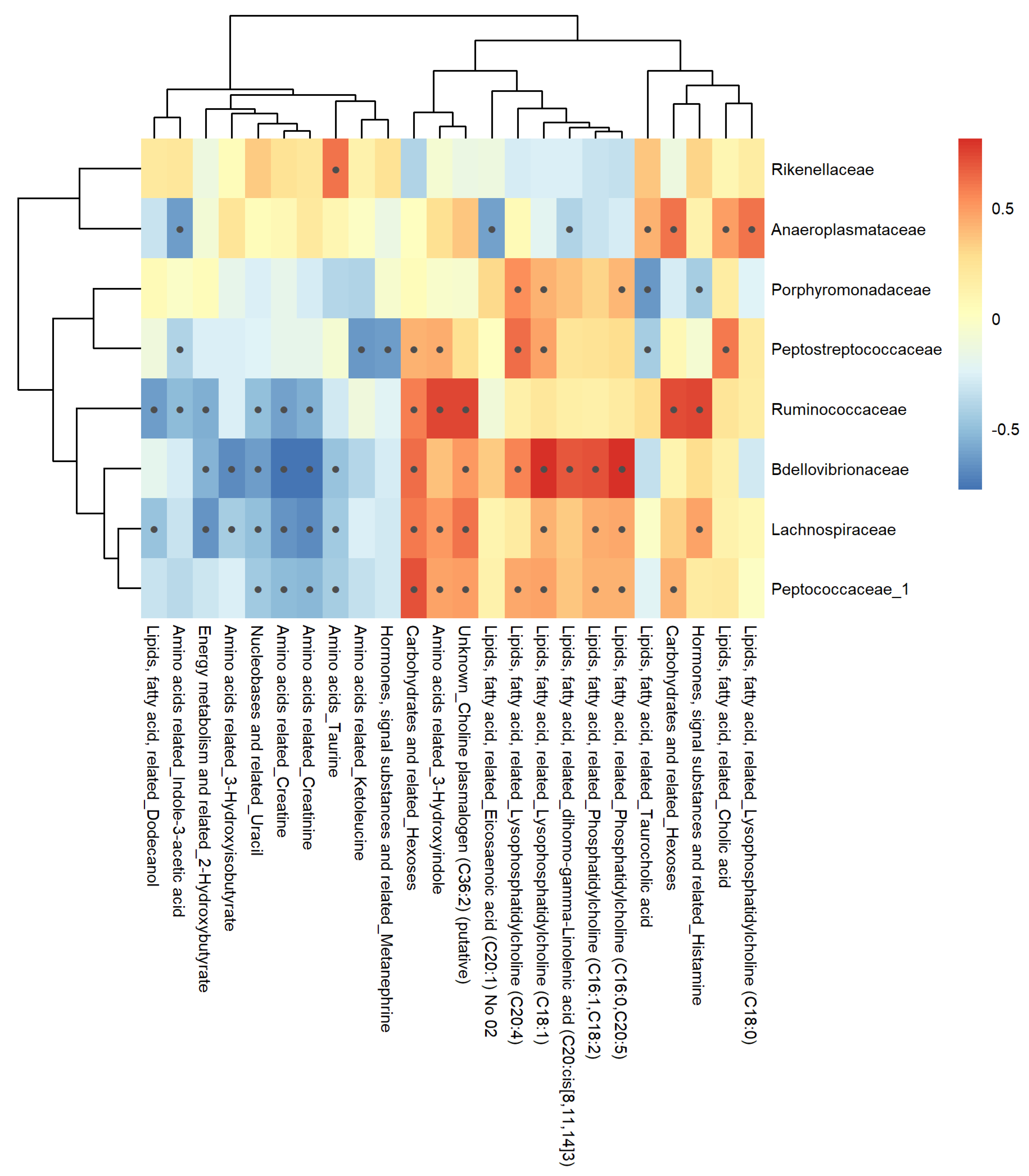
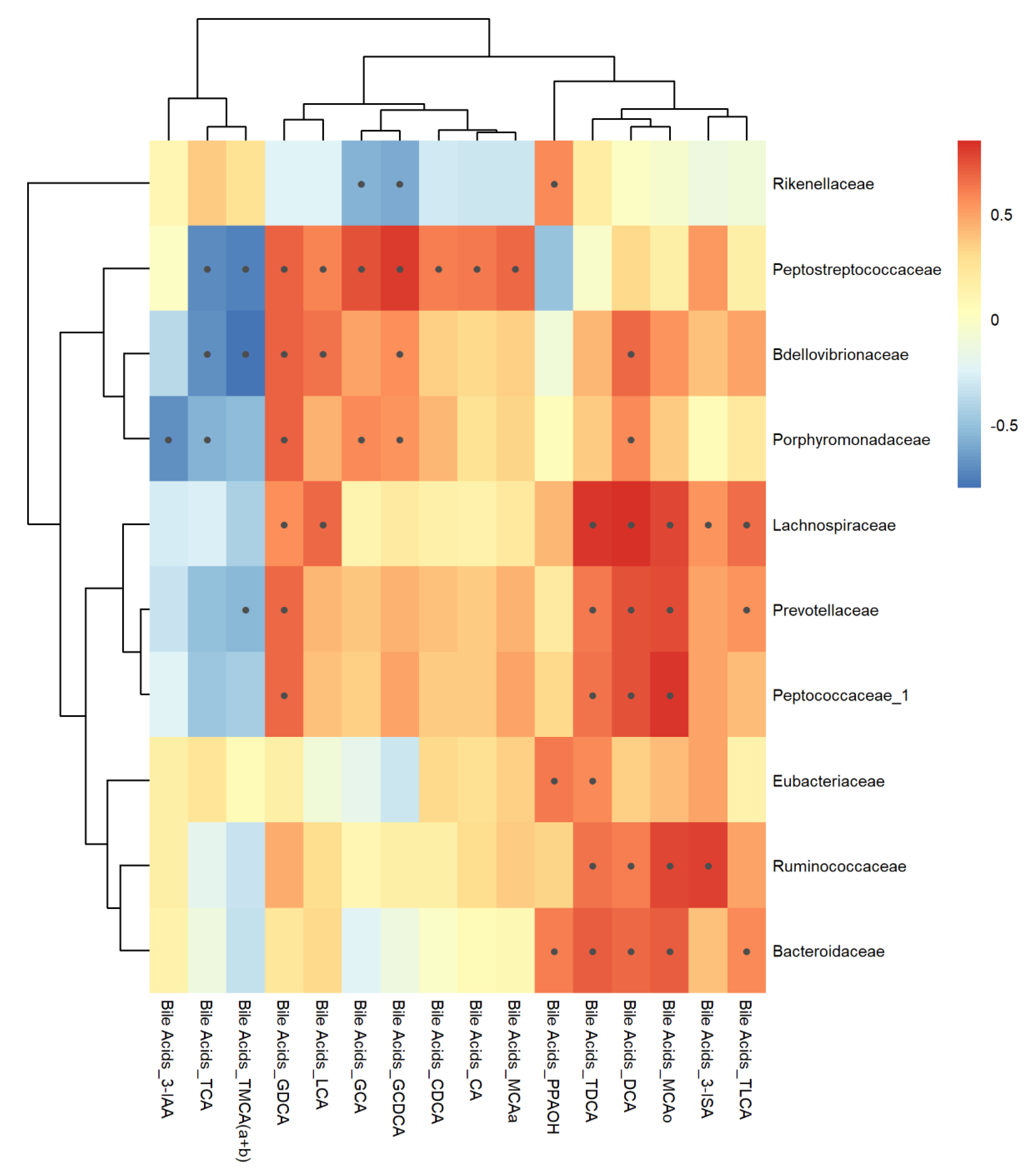
3.7. Correlation Analysis
3.7.1. Feces Matrix
3.7.2. Plasma Matrix
4. Discussion
4.1. Microbiome Analysis
4.2. Metabolome Analysis
4.3. Correlation Analysis
4.3.1. Feces Metabolome-Microbiome
4.3.2. Plasma Metabolome-Microbiome
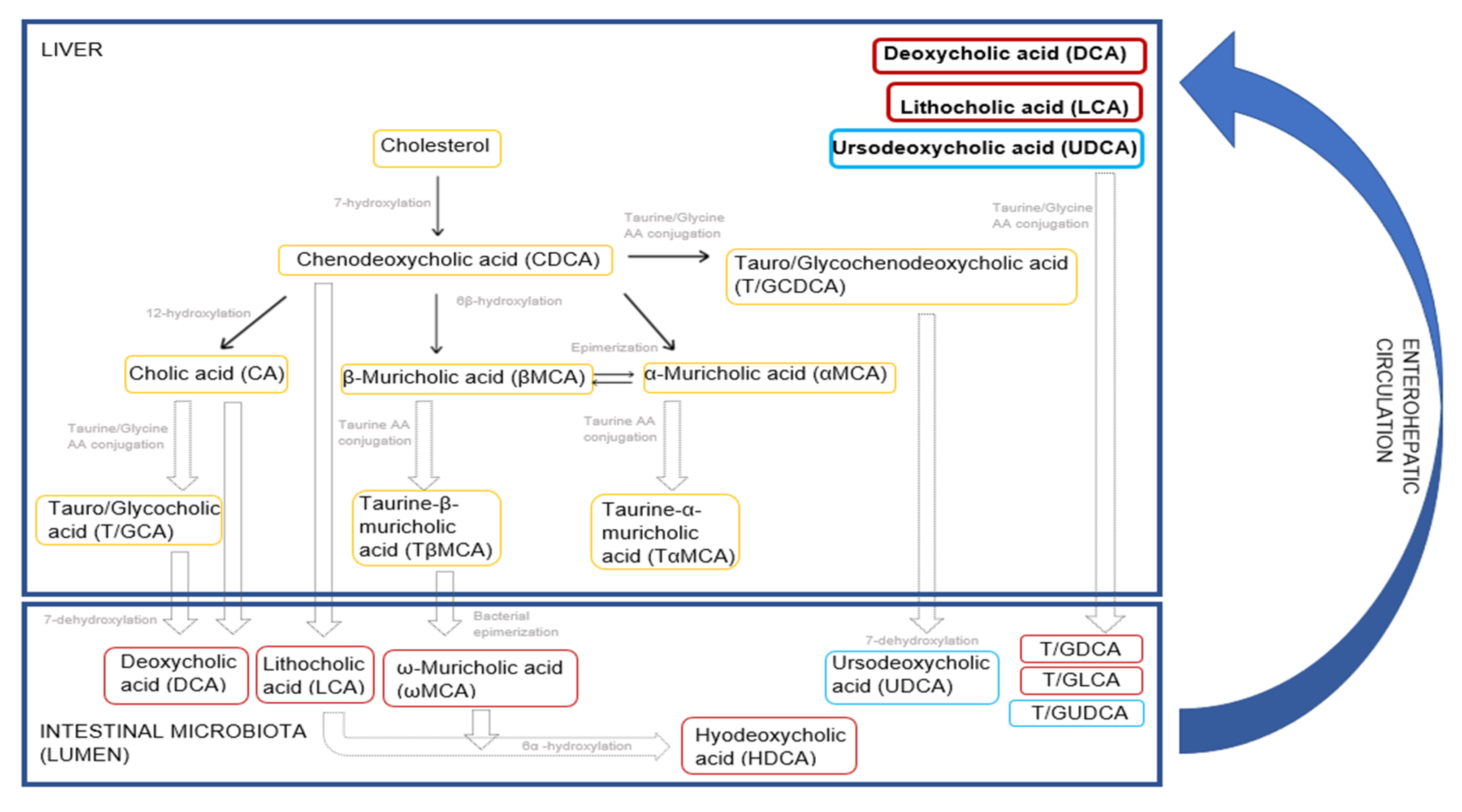
4.4. Bile Acids
4.4.1. Clindamycin Hydrochloride Treatment
4.4.2. Vancomycin Treatment
4.4.3. Sparfloxacin Treatment
4.4.4. Roxithromycin Treatment
4.4.5. Streptomycin Sulfate Treatment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, D.; Chen, H.; Mao, B.; Yang, Q.; Zhao, J.; Gu, Z.; Zhang, H.; Chen, Y.Q.; Chen, W. Microbial Biogeography and Core Microbiota of the Rat Digestive Tract. Sci. Rep. 2017, 7, 45840. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic Analysis of the Human Distal Gut Microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nat. Cell Biol. 2019, 570, 462–467. [Google Scholar] [CrossRef]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut Reactions: Breaking Down Xenobiotic–Microbiome Interactions. Pharmacol. Rev. 2019, 71, 198–224. [Google Scholar] [CrossRef]
- ECETOC. Microbiome Expert Workshop Report (Porto, 8–9 July 2019); 2078-7219-036; ECETOC: Brussels, Belgium, 2020. [Google Scholar]
- Bourassa, M.W.; Alim, I.; Bultman, S.J.; Ratan, R.R. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci. Lett. 2016, 625, 56–63. [Google Scholar] [CrossRef]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nat. Cell Biol. 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chen, M.; Li, Y.; Wang, Y.; Wei, L.; Liao, Z.; Wang, M.; Ma, F.; Liao, Q.; Xie, Z. Modulation of Gut Microbiome Composition and Function in Experimental Colitis Treated with Sulfasalazine. Front. Microbiol. 2017, 8, 1703. [Google Scholar] [CrossRef] [PubMed]
- Ghaisas, S.; Maher, J.; Kanthasamy, A. Gut microbiome in health and disease: Linking the microbiome–gut–brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 2016, 158, 52–62. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef]
- Anders, M. Metabolism of drugs by the kidney. Kidney Int. 1980, 18, 636–647. [Google Scholar] [CrossRef]
- Behr, C.; Kamp, H.; Fabian, E.; Krennrich, G.; Mellert, W.; Peter, E.; Strauss, V.; Walk, T.; Rietjens, I.M.C.M.; Van Ravenzwaay, B. Gut microbiome-related metabolic changes in plasma of antibiotic-treated rats. Arch. Toxicol. 2017, 91, 3439–3454. [Google Scholar] [CrossRef]
- Behr, C.; Ramírez-Hincapié, S.; Cameron, H.; Strauss, V.; Walk, T.; Herold, M.; Beekmann, K.; Rietjens, I.; Van Ravenzwaay, B. Impact of lincosamides antibiotics on the composition of the rat gut microbiota and the metabolite profile of plasma and feces. Toxicol. Lett. 2018, 296, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Behr, C.; Slopianka, M.; Haake, V.; Strauss, V.; Sperber, S.; Kamp, H.; Walk, T.; Beekmann, K.; Rietjens, I.; Van Ravenzwaay, B. Analysis of metabolome changes in the bile acid pool in feces and plasma of antibiotic-treated rats. Toxicol. Appl. Pharmacol. 2019, 363, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Behr, C.; Sperber, S.; Jiang, X.; Strauss, V.; Kamp, H.; Walk, T.; Herold, M.; Beekmann, K.; Rietjens, I.; van Ravenzwaay, B. Microbiome-related metabolite changes in gut tissue, cecum content and feces of rats treated with antibiotics. Toxicol. Appl. Pharmacol. 2018, 355, 198–210. [Google Scholar] [CrossRef]
- Van Ravenzwaay, B.; Herold, M.; Kamp, H.; Kapp, M.; Fabian, E.; Looser, R.; Krennrich, G.; Mellert, W.; Prokoudine, A.; Strauss, V.; et al. Metabolomics: A tool for early detection of toxicological effects and an opportunity for biology based grouping of chemicals—From QSAR to QBAR. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 746, 144–150. [Google Scholar] [CrossRef]
- Van Ravenzwaay, B.; Sperber, S.; Lemke, O.; Fabian, E.; Faulhammer, F.; Kamp, H.; Mellert, W.; Strauss, V.; Strigun, A.; Peter, E.; et al. Metabolomics as read-across tool: A case study with phenoxy herbicides. Regul. Toxicol. Pharmacol. 2016, 81, 288–304. [Google Scholar] [CrossRef]
- Van Ravenzwaay, B.; Cunha, G.C.-P.; Leibold, E.; Looser, R.; Mellert, W.; Prokoudine, A.; Walk, T.; Wiemer, J. The use of metabolomics for the discovery of new biomarkers of effect. Toxicol. Lett. 2007, 172, 21–28. [Google Scholar] [CrossRef]
- De Bruijn, V.; Behr, C.; Sperber, S.; Walk, T.; Ternes, P.; Slopianka, M.; Haake, V.; Beekmann, K.; Van Ravenzwaay, B. Antibiotic-Induced Changes in Microbiome-Related Metabolites and Bile Acids in Rat Plasma. Metabolites 2020, 10, 242. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio: Boston, MA, USA, 2020. [Google Scholar]
- Troyanskaya, O.G.; Cantor, M.; Sherlock, G.; Brown, P.O.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. PHYLOSEQ: A Bioconductor Package for Handling and Analysis of High-Throughput Phylogenetic Sequence Data. In Biocomputing; Springer: Berlin/Heidelberg, Germany, 2012; pp. 235–246. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R. Package ‘Vegan’. Community Ecology Package, Version 2; R Core Team: Vienna, Austria, 2013. [Google Scholar]
- Liu, C.M.; Bs, K.S.; Nordstrom, L.; Dwan, M.G.; Moss, O.L.; Contente-Cuomo, T.L.; Keim, P.; Price, L.B.; Lane, A.P.; Bs, M.G.D.; et al. Medical therapy reduces microbiota diversity and evenness in surgically recalcitrant chronic rhinosinusitis. Int. Forum Allergy Rhinol. 2013, 3, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Fraumene, C.; Manghina, V.; Cadoni, E.; Marongiu, F.; Abbondio, M.; Serra, M.; Palomba, A.; Tanca, A.; Laconi, E.; Uzzau, S. Caloric restriction promotes rapid expansion and long-lasting increase of Lactobacillus in the rat fecal microbiota. Gut Microbes 2017, 9, 104–114. [Google Scholar] [CrossRef]
- Verdier, L.; Bertho, G.; Gharbi-Benarous, J.; Girault, J.-P. Lincomycin and clindamycin conformations. A fragment shared by macrolides, ketolides and lincosamides determined from TRNOE ribosome-bound conformations. Bioorganic Med. Chem. 2000, 8, 1225–1243. [Google Scholar] [CrossRef]
- Zhou, G.; An, Z.; Zhao, W.; Hong, Y.; Xin, H.; Ning, X.; Wang, J. Sex differences in outcomes after stroke among patients with low total cholesterol levels: A large hospital-based prospective study. Biol. Sex Differ. 2016, 7, 62. [Google Scholar] [CrossRef][Green Version]
- Kim, Y.S.; Unno, T.; Kim, B.-Y.; Park, M.-S. Sex Differences in Gut Microbiota. World J. Men’s Health 2020, 38, 48–60. [Google Scholar] [CrossRef]
- Wishart, D.S. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Bao, H.-D.; Pang, M.-D.; Olaniran, A.; Zhang, X.-H.; Zhang, H.; Zhou, Y.; Sun, L.-C.; Schmidt, S.; Wang, R. Alterations in the diversity and composition of mice gut microbiota by lytic or temperate gut phage treatment. Appl. Microbiol. Biotechnol. 2018, 102, 10219–10230. [Google Scholar] [CrossRef]
- Zhang, C.; Li, X.; Liu, L.; Gao, L.; Ou, S.; Luo, J.; Peng, X. Roxithromycin regulates intestinal microbiota and alters colonic epithelial gene expression. Appl. Microbiol. Biotechnol. 2018, 102, 9303–9316. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wang, S.; Jia, W. Calorie restriction and its impact on gut microbial composition and global metabolism. Front. Med. 2018, 12, 634–644. [Google Scholar] [CrossRef]
- Zeng, H.; Grapov, D.; Jackson, M.I.; Fahrmann, J.F.; Fiehn, O.; Combs, G.F. Integrating Multiple Analytical Datasets to Compare Metabolite Profiles of Mouse Colonic-Cecal Contents and Feces. Metabolites 2015, 5, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Ninnes, C.E.; Waas, J.R.; Ling, N.; Nakagawa, S.; Banks, J.C.; Bell, D.G.; Bright, A.; Carey, P.W.; Chandler, J.; Hudson, Q.J.; et al. Comparing plasma and faecal measures of steroid hormones in Adelie penguins Pygoscelis adeliae. J. Comp. Physiol. B 2009, 180, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Fujisaka, S.; Avila-Pacheco, J.; Soto, M.; Kostic, A.; Dreyfuss, J.M.; Pan, H.; Ussar, S.; Altindis, E.; Li, N.; Bry, L.; et al. Diet, Genetics, and the Gut Microbiome Drive Dynamic Changes in Plasma Metabolites. Cell Rep. 2018, 22, 3072–3086. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.; Citron, D.M.; Gerardo, S.H.; Hudspeth, M.; Merriam, C.V. Comparative in vitro activities of DU-6859a, levofloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against 387 aerobic and anaerobic bite wound isolates. Antimicrob. Agents Chemother. 1997, 41, 1193–1195. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bahar, H.; Torun, M.M.; Demirci, M.; Kocazeybek, B. Antimicrobial Resistance and β-Lactamase Production of Clinical Isolates of Prevotella and Porphyromonas Species. Chemotherapy 2005, 51, 9–14. [Google Scholar] [CrossRef]
- Dai, Z.-L. Amino acid metabolism in intestinal bacteria: Links between gut ecology and host health. Front. Biosci. 2011, 16, 1768–1786. [Google Scholar] [CrossRef]
- Alkhalaf, L.M.; Ryan, K.S. Biosynthetic Manipulation of Tryptophan in Bacteria: Pathways and Mechanisms. Chem. Biol. 2015, 22, 317–328. [Google Scholar] [CrossRef]
- Rikitake, K.; Oka, I.; Ando, M.; Yoshimoto, T.; Tsuru, D. Creatinine Amidohydrolase (Creatininase) from Pseudomonas putida: ePurification and Some Properties1. J. Biochem. 1979, 86, 1109–1117. [Google Scholar] [CrossRef]
- Idris, E.E.; Iglesias, D.J.; Talon, M.; Borriss, R. Tryptophan-Dependent Production of Indole-3-Acetic Acid (IAA) Affects Level of Plant Growth Promotion by Bacillus amyloliquefaciens FZB42. Mol. Plant-Microbe Interact. 2007, 20, 619–626. [Google Scholar] [CrossRef]
- Sanguinetti, E.; Collado, M.C.; Marrachelli, V.G.; Monleon, D.; Selma-Royo, M.; Pardo-Tendero, M.M.; Burchielli, S.; Iozzo, P. Microbiome-metabolome signatures in mice genetically prone to develop dementia, fed a normal or fatty diet. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Apper, E.; Privet, L.; Taminiau, B.; Le Bourgot, C.; Svilar, L.; Martin, J.-C.; Diez, M. Relationships between Gut Microbiota, Metabolome, Body Weight, and Glucose Homeostasis of Obese Dogs Fed with Diets Differing in Prebiotic and Protein Content. Microorganisms 2020, 8, 513. [Google Scholar] [CrossRef]
- Steinway, S.N.; Biggs, M.B.; Jr, T.P.L.; Papin, J.A.; Albert, R. Inference of Network Dynamics and Metabolic Interactions in the Gut Microbiome. PLoS Comput. Biol. 2015, 11, e1004338. [Google Scholar] [CrossRef]
- Zhao, Z.-H.; Xin, F.-Z.; Xue, Y.; Hu, Z.; Han, Y.; Ma, F.; Zhou, D.; Liu, X.-L.; Cui, A.; Liu, Z.; et al. Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Jazani, N.H.; Savoj, J.; Lustgarten, M.; Lau, W.L.; Vaziri, N.D. Impact of Gut Dysbiosis on Neurohormonal Pathways in Chronic Kidney Disease. Diseases 2019, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xie, G.; Zhao, A.; Zhao, L.; Yao, C.; Chiu, N.H.L.; Zhou, Z.; Bao, Y.; Jia, W.; Nicholson, J.K.; et al. The Footprints of Gut Microbial–Mammalian Co-Metabolism. J. Proteome Res. 2011, 10, 5512–5522. [Google Scholar] [CrossRef] [PubMed]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Martin, F.J.; Dumas, M.; Wang, Y.; Legido-Quigley, C.; Yap, I.K.S.; Tang, H.; Zirah, S.; Murphy, G.M.; Cloarec, O.; Lindon, J.C.; et al. A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol. Syst. Biol. 2007, 3, 112. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Baccetto, R.L.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Number | Treatment | Low Dose (mg/kg bw/day) | High Dose (mg/kg bw/day) | Caging | Form of Preparation | Class of Antibiotics |
|---|---|---|---|---|---|---|
| 1–4 | Control diet | - | - | Grouped (5) | - | - |
| 1 | Vancomycin | 50 | 400 | Grouped (5) | in ultra-pure water | Glycopeptides |
| 1 | Streptomycin sulfate | 100 | 450 | Grouped (5) | in water containing 0.5% CMC a | Aminoglycosides |
| 1 | Roxithromycin | 200 | 600 | Grouped (5) | in water containing 0.5% CMC a | Macrolides |
| 2 | Sparfloxacin | 200 | 600 | Grouped (5) | in water containing 0.5% CMC a | Fluoroquinolones |
| 3 | Restricted diet (−20%) | - | - | Single (1) | - | - |
| 4 | Clindamycin hydrochloride | 200 | 600 | Grouped (5) | in ultra-pure water | Lincosamides |
| 4 | Lincomycin hydrochloride | 300 | 10000 | Grouped (5) | in water containing 0.5% CMC a | Lincosamides |
| Metabolite Name | Analyte Name | Feces | Plasma | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F | M | F 7d | F 14d | F 28d | M 7d | M 14d | M 28d | ||
| Cholate | CA | 18.64 | 19.90 | 0.02 | 0.03 | 0.01 | 0.01 | 0.03 | 0.01 |
| Chenodeoxycholate (chenodeoxycholic acid) | CDCA | 0.30 | 0.08 | 0.02 | 0.03 | 0.04 | 0.02 | 0.10 | 0.02 |
| Deoxycholate (deoxycholic acid) | DCA | 0.00 | 0.00 | 0.35 | 0.00 | 0.01 | 0.10 | 0.36 | 0.01 |
| Glycocholate, glycocholic acid | GCA | 1.72 | 0.47 | 0.44 | 0.30 | 0.21 | 0.22 | 0.35 | 0.16 |
| Glycochenodeoxycholate (glycochenodeoxycholic acid) | GCDCA | 2.67 | 0.09 | 0.56 | 0.40 | 0.76 | 0.06 | 0.08 | 0.06 |
| Glycodeoxycholate, -cholic acid | GDCA | 0.58 | 0.04 | 0.07 | 0.01 | 0.01 | 0.02 | ||
| Glycolithocholic aicd | GLCA | 0.52 | 0.44 | 0.54 | 0.26 | 0.68 | 0.12 | 0.79 | 0.36 |
| Glycoursodeoxycholic acid | GUDCA | 2.82 | 2.25 | 0.11 | 1.03 | 0.97 | 0.30 | ||
| Hyodeoxycholate, hyodeoxycholic acid | HDCA | 0.00 | 0.00 | 0.01 | 0.01 | 0.00 | 0.01 | 0.00 | |
| Lithocholate, lithocholic acid | LCA | 0.01 | 0.01 | 1.00 | 0.53 | 1.55 | 0.35 | ||
| Muricholic acid (alpha) | MCAa | 0.59 | 0.16 | 0.03 | 0.06 | 0.06 | 0.01 | 0.02 | 0.01 |
| Muricholic acid (beta) | MCAb | 2.36 | 0.87 | 0.11 | 0.25 | 0.39 | 0.15 | 0.31 | 0.39 |
| Muricholic acid (omega) | MCAo | 0.06 | 0.01 | 0.00 | 0.01 | 0.04 | 0.01 | 0.00 | 0.01 |
| Taurocholate, taurocholic acid | TCA | 47.42 | 90.80 | 1.34 | 1.01 | 1.63 | 4.68 | 2.94 | 4.56 |
| Taurochenodeoxycholate | TCDCA | 35.03 | 13.64 | 1.38 | 0.99 | 1.54 | 2.46 | 1.46 | 1.25 |
| Taurodeoxycholate, -cholic acid | TDCA | 0.25 | 0.15 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | 0.05 |
| Taurolithocholic aicd | TLCA | 0.15 | 0.34 | 0.04 | 0.04 | 0.19 | 0.14 | 0.19 | |
| Tauromuricholic acid (a + b) | TMCA (a + b) | 155.83 | 192.72 | 1.96 | 1.41 | 2.38 | 4.97 | 2.57 | 3.99 |
| Tauroursodeoxycholic acid | TUDCA | 22.61 | 28.27 | 6.92 | 0.20 | 0.57 | 0.74 | ||
| Ursodeoxycholate, Ursodeoxycholic acid, Ursodiol | UDCA | 0.02 | 0.03 | 0.05 | 0.27 | 0.01 | 0.01 | 0.30 | 0.02 |
| Metabolite Name | Analyte Name | Feces | Plasma | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F | M | F 7d | F 14d | F 28d | M 7d | M 14d | M 28d | ||
| Cholate | CA | 19.45 | 38.66 | 0.18 | 0.60 | 0.14 | 0.10 | 0.13 | 0.13 |
| Chenodeoxycholate (chenodeoxycholic acid) | CDCA | 4.30 | 0.99 | 0.18 | 0.36 | 0.05 | 0.10 | 0.16 | 0.18 |
| Deoxycholate (deoxycholic acid) | DCA | 0.00 | 0.00 | 0.01 | 0.42 | 0.01 | 0.00 | 0.18 | 0.00 |
| Glycocholate, glycocholic acid | GCA | 2.67 | 1.45 | 0.46 | 1.54 | 0.73 | 0.39 | 0.36 | 0.18 |
| Glycochenodeoxycholate (glycochenodeoxycholic acid) | GCDCA | 1.01 | 1.54 | 0.39 | 0.87 | 0.64 | 0.30 | 0.27 | 0.42 |
| Glycodeoxycholate, -cholic acid | GDCA | 0.32 | 0.34 | 0.02 | 0.07 | 0.03 | 0.01 | 0.00 | 0.05 |
| Glycolithocholic aicd | GLCA | 0.24 | 0.86 | 0.56 | 0.21 | 0.20 | 0.48 | 1.65 | 4.39 |
| Glycoursodeoxycholic acid | GUDCA | 1.44 | 1.44 | 1.65 | 1.18 | 3.31 | 0.43 | 16.34 | 10.13 |
| Hyodeoxycholate, hyodeoxycholic acid | HDCA | 0.00 | 0.00 | 0.01 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 |
| Lithocholate, lithocholic acid | LCA | 0.01 | 0.07 | 0.12 | 10.74 | 0.15 | 0.35 | 27.95 | 0.19 |
| Muricholic acid (alpha) | MCAa | 0.44 | 0.71 | 0.21 | 0.44 | 0.10 | 0.13 | 0.14 | 0.23 |
| Muricholic acid (beta) | MCAb | 0.50 | 0.53 | 0.54 | 1.01 | 1.35 | 0.17 | 0.18 | 0.38 |
| Muricholic acid (omega) | MCAo | 0.02 | 0.03 | 0.05 | 0.01 | 0.07 | 0.03 | 0.01 | 0.02 |
| Taurocholate, taurocholic acid | TCA | 4.18 | 7.39 | 2.04 | 6.99 | 3.83 | 2.47 | 2.71 | 2.62 |
| Taurochenodeoxycholate | TCDCA | 0.99 | 3.04 | 1.87 | 2.42 | 2.64 | 2.69 | 3.18 | 2.97 |
| Taurodeoxycholate, -cholic acid | TDCA | 0.16 | 0.09 | 0.01 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 |
| Taurolithocholic aicd | TLCA | 0.42 | 0.03 | 0.04 | 0.03 | 0.12 | 0.18 | 1.58 | |
| Tauromuricholic acid (a + b) | TMCA (a + b) | 2.86 | 3.89 | 3.10 | 4.02 | 3.59 | 1.93 | 2.71 | 3.11 |
| Tauroursodeoxycholic acid | TUDCA | 0.54 | 2.93 | 3.61 | 1.82 | 1.23 | 0.79 | ||
| Ursodeoxycholate, Ursodeoxycholic acid, Ursodiol | UDCA | 0.21 | 0.06 | 0.29 | 0.13 | 0.12 | 0.23 | ||
| Metabolite Name | Analyte Name | Feces | Plasma | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F | M | F 7d | F 14d | F 28d | M 7d | M 14d | M 28d | ||
| Cholate | CA | 27.39 | 22.73 | 1.08 | 0.46 | 0.32 | 0.11 | 0.91 | 0.90 |
| Chenodeoxycholate (chenodeoxycholic acid) | CDCA | 0.52 | 0.32 | 0.17 | 0.37 | 0.04 | 0.28 | 0.94 | |
| Deoxycholate (deoxycholic acid) | DCA | 0.01 | 0.00 | 0.09 | 0.02 | 0.08 | 0.01 | 0.18 | 0.03 |
| Glycocholate, glycocholic acid | GCA | 1.88 | 1.57 | 1.02 | 0.53 | 2.05 | 0.42 | 0.59 | 0.71 |
| Glycochenodeoxycholate (glycochenodeoxycholic acid) | GCDCA | 1.25 | 2.93 | 0.51 | 0.34 | 1.61 | 0.14 | 0.13 | 0.40 |
| Glycodeoxycholate, -cholic acid | GDCA | 2.70 | 0.08 | 0.25 | 0.51 | 0.02 | 0.01 | 0.12 | |
| Glycolithocholic aicd | GLCA | 8.22 | 1.33 | 0.90 | 0.71 | 0.24 | 1.93 | ||
| Glycoursodeoxycholic acid | GUDCA | 0.96 | 1.61 | 0.33 | 1.30 | ||||
| Hyodeoxycholate, hyodeoxycholic acid | HDCA | 0.01 | 0.00 | 0.06 | 0.01 | 0.00 | 0.00 | ||
| Lithocholate, lithocholic acid | LCA | 0.24 | 0.01 | 0.12 | 0.01 | 13.09 | 0.18 | 9.66 | 0.31 |
| Muricholic acid (alpha) | MCAa | 0.58 | 0.16 | 0.67 | 0.24 | 0.57 | 0.05 | 0.38 | 0.73 |
| Muricholic acid (beta) | MCAb | 0.33 | 0.37 | 1.46 | 1.21 | 0.31 | 0.25 | 1.87 | 1.10 |
| Muricholic acid (omega) | MCAo | 0.01 | 0.02 | 0.20 | 0.01 | 0.01 | 0.01 | 0.00 | |
| Taurocholate, taurocholic acid | TCA | 4.95 | 8.29 | 2.61 | 3.25 | 1.73 | 7.96 | 5.32 | 3.09 |
| Taurochenodeoxycholate | TCDCA | 2.29 | 4.99 | 1.21 | 1.44 | 1.77 | 3.85 | 3.63 | 2.08 |
| Taurodeoxycholate, -cholic acid | TDCA | 0.30 | 0.18 | 0.09 | 0.21 | 0.03 | 0.11 | 0.03 | 0.04 |
| Taurolithocholic aicd | TLCA | 0.27 | 0.75 | 1.11 | 0.06 | 0.28 | 0.33 | 0.22 | |
| Tauromuricholic acid (a + b) | TMCA (a + b) | 7.08 | 16.86 | 1.72 | 1.73 | 1.88 | 4.25 | 4.95 | 2.27 |
| Tauroursodeoxycholic acid | TUDCA | 2.12 | 9.35 | 10.27 | 7.76 | 1.82 | |||
| Ursodeoxycholate, Ursodeoxycholic acid, Ursodiol | UDCA | 0.25 | 0.01 | 0.36 | 0.56 | 0.23 | 0.13 | 1.20 | 1.17 |
| Metabolite Name | Analyte Name | Feces | Plasma | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F | M | F 7d | F 14 d | F 28 d | M 7d | M 14 d | M 28 d | ||
| Cholate | CA | 0.52 | 0.55 | 0.04 | 0.05 | 0.13 | 0.00 | 0.00 | 0.02 |
| Chenodeoxycholate (chenodeoxycholic acid) | CDCA | 0.90 | 0.02 | 0.06 | 0.08 | 0.02 | 0.01 | 0.05 | |
| Deoxycholate (deoxycholic acid) | DCA | 0.49 | 0.70 | 0.75 | 0.58 | 0.86 | 0.15 | 0.11 | 0.20 |
| Glycocholate, glycocholic acid | GCA | 0.15 | 0.23 | 0.42 | 0.30 | 0.10 | 0.25 | 0.10 | 0.08 |
| Glycochenodeoxycholate (glycochenodeoxycholic acid) | GCDCA | 0.42 | 0.72 | 0.39 | 0.36 | 0.17 | 0.38 | 0.10 | 0.08 |
| Glycodeoxycholate, -cholic acid | GDCA | 0.49 | 0.51 | 0.37 | 0.07 | 0.14 | 0.05 | 0.07 | |
| Glycolithocholic aicd | GLCA | 0.41 | 1.63 | 0.28 | 0.27 | 0.49 | 0.33 | 0.93 | 0.66 |
| Glycoursodeoxycholic acid | GUDCA | 1.44 | 1.82 | 0.59 | 1.13 | 0.46 | 1.45 | 0.63 | |
| Hyodeoxycholate, hyodeoxycholic acid | HDCA | 0.06 | 0.03 | 0.10 | 0.04 | 0.10 | 0.01 | 0.01 | 0.01 |
| Lithocholate, lithocholic acid | LCA | 0.23 | 0.35 | 0.71 | 0.46 | 0.72 | 0.64 | 6.52 | 0.50 |
| Muricholic acid (alpha) | MCAa | 0.73 | 0.62 | 0.22 | 0.19 | 0.11 | 0.08 | 0.02 | 0.05 |
| Muricholic acid (beta) | MCAb | 1.33 | 0.84 | 0.71 | 1.30 | 2.28 | 0.16 | 0.05 | 0.22 |
| Muricholic acid (omega) | MCAo | 0.77 | 0.60 | 1.19 | 0.60 | 1.47 | 0.74 | 0.36 | 0.42 |
| Taurocholate, taurocholic acid | TCA | 2.41 | 1.58 | 2.59 | 5.55 | 3.14 | 3.35 | 2.19 | 3.77 |
| Taurochenodeoxycholate | TCDCA | 1.42 | 1.54 | 1.69 | 1.48 | 1.84 | 2.07 | 2.02 | 2.28 |
| Taurodeoxycholate, -cholic acid | TDCA | 9.42 | 4.61 | 2.29 | 3.11 | 1.89 | 2.55 | 1.19 | 2.06 |
| Taurolithocholic aicd | TLCA | 3.26 | 3.13 | 0.74 | 0.75 | 1.39 | 1.05 | 0.68 | 0.61 |
| Tauromuricholic acid (a + b) | TMCA (a + b) | 10.14 | 7.97 | 1.94 | 2.94 | 2.37 | 3.62 | 2.67 | 3.02 |
| Tauroursodeoxycholic acid | TUDCA | 3.42 | 2.20 | 9.03 | 1.23 | 1.24 | 0.55 | ||
| Ursodeoxycholate, Ursodeoxycholic acid, Ursodiol | UDCA | 0.11 | 0.06 | 0.53 | 0.03 | 0.05 | 0.01 | ||
| Metabolite Name | Analyte Name | Feces | Plasma | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F | M | F | M | F | M | F | M | ||
| Cholate | CA | 8.33 | 3.68 | 0.49 | 0.97 | 0.14 | 0.71 | 0.01 | 0.24 |
| Chenodeoxycholate (chenodeoxycholic acid) | CDCA | 17.60 | 0.56 | 0.43 | 1.65 | 0.17 | 0.64 | 0.02 | 0.37 |
| Deoxycholate (deoxycholic acid) | DCA | 1.15 | 1.56 | 0.67 | 0.68 | 1.22 | 0.62 | 0.15 | 0.31 |
| Glycocholate, glycocholic acid | GCA | 1.14 | 0.29 | 3.56 | 0.72 | 0.58 | 1.53 | 0.35 | 0.26 |
| Glycochenodeoxycholate (glycochenodeoxycholic acid) | GCDCA | 0.61 | 0.70 | 1.28 | 0.93 | 0.98 | 0.41 | 0.10 | 0.31 |
| Glycodeoxycholate, -cholic acid | GDCA | 0.39 | 0.21 | 1.54 | 1.02 | 0.49 | 0.23 | 0.05 | 0.10 |
| Glycolithocholic aicd | GLCA | 0.35 | 1.41 | 0.46 | 0.27 | 0.23 | 0.39 | 0.25 | 0.80 |
| Glycoursodeoxycholic acid | GUDCA | 0.89 | 0.50 | 1.65 | 0.61 | 1.00 | 1.06 | 1.24 | 0.89 |
| Hyodeoxycholate, hyodeoxycholic acid | HDCA | 0.20 | 0.04 | 0.18 | 0.25 | 0.34 | 0.03 | 0.01 | 0.03 |
| Lithocholate, lithocholic acid | LCA | 0.35 | 0.89 | 0.68 | 0.39 | 0.67 | 0.69 | 0.35 | 0.43 |
| Muricholic acid (alpha) | MCAa | 2.95 | 2.43 | 0.45 | 1.03 | 0.42 | 1.11 | 0.04 | 0.32 |
| Muricholic acid (beta) | MCAb | 1.07 | 2.79 | 1.89 | 1.81 | 0.77 | 1.89 | 0.53 | 0.87 |
| Muricholic acid (omega) | MCAo | 2.11 | 1.77 | 2.17 | 1.46 | 2.51 | 3.19 | 0.36 | 0.78 |
| Taurocholate, taurocholic acid | TCA | 2.07 | 1.13 | 1.73 | 2.97 | 2.05 | 4.50 | 3.28 | 2.67 |
| Taurochenodeoxycholate | TCDCA | 1.17 | 1.15 | 1.22 | 1.35 | 2.61 | 2.54 | 2.33 | 2.72 |
| taurodeoxycholate, -cholic acid | TDCA | 2.25 | 0.62 | 1.66 | 1.62 | 2.13 | 1.68 | 1.38 | 1.48 |
| Taurolithocholic aicd | TLCA | 0.60 | 1.68 | 1.11 | 0.83 | 1.05 | 0.66 | 0.38 | 0.47 |
| Tauromuricholic acid (a + b) | TMCA (a + b) | 1.86 | 1.79 | 1.60 | 2.44 | 2.59 | 3.01 | 2.64 | 2.64 |
| Tauroursodeoxycholic acid | TUDCA | 1.79 | 1.04 | 2.83 | 1.98 | 0.63 | |||
| ursodeoxycholate, ursodeoxycholic acid, ursodiol | UDCA | 1.65 | 0.16 | 0.20 | 1.60 | 0.17 | 0.39 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murali, A.; Giri, V.; Cameron, H.J.; Behr, C.; Sperber, S.; Kamp, H.; Walk, T.; van Ravenzwaay, B. Elucidating the Relations between Gut Bacterial Composition and the Plasma and Fecal Metabolomes of Antibiotic Treated Wistar Rats. Microbiol. Res. 2021, 12, 82-122. https://doi.org/10.3390/microbiolres12010008
Murali A, Giri V, Cameron HJ, Behr C, Sperber S, Kamp H, Walk T, van Ravenzwaay B. Elucidating the Relations between Gut Bacterial Composition and the Plasma and Fecal Metabolomes of Antibiotic Treated Wistar Rats. Microbiology Research. 2021; 12(1):82-122. https://doi.org/10.3390/microbiolres12010008
Chicago/Turabian StyleMurali, Aishwarya, Varun Giri, Hunter James Cameron, Christina Behr, Saskia Sperber, Hennicke Kamp, Tilmann Walk, and Bennard van Ravenzwaay. 2021. "Elucidating the Relations between Gut Bacterial Composition and the Plasma and Fecal Metabolomes of Antibiotic Treated Wistar Rats" Microbiology Research 12, no. 1: 82-122. https://doi.org/10.3390/microbiolres12010008
APA StyleMurali, A., Giri, V., Cameron, H. J., Behr, C., Sperber, S., Kamp, H., Walk, T., & van Ravenzwaay, B. (2021). Elucidating the Relations between Gut Bacterial Composition and the Plasma and Fecal Metabolomes of Antibiotic Treated Wistar Rats. Microbiology Research, 12(1), 82-122. https://doi.org/10.3390/microbiolres12010008