_Rachiotis.png)
COVID-19 and Parasitic Co-Infection: A Hypothetical Link to Pulmonary Vascular Disease

 , ,
, ,  , ,
, ,  ,
, 

Abstract
1. Introduction
2. Methods
3. COVID-19 and Parasitic Co-Infection
3.1. Schistosomiasis and Other Helminthic Diseases
3.2. HIV and Viral Infections Like Other Human Herpesviruses
3.3. Tuberculosis and Other Bacterial Infections
3.4. Pulmonary Aspergillosis and Other Fungal Infections
3.5. Gaps in Evidence
4. A Multidisciplinary Diagnostic and Management Approach of PVDs Related to COVID-19 Co-Infections Is Important
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
References
- Oliveira, S.D.; Almodóvar, S.; Butrous, G.; De Jesus Perez, V.; Fabro, A.; Graham, B.B.; Mocumbi, A.; Nyasulu, P.S.; Tura-Ceide, O.; Oliveira, R.K.F.; et al. Infection and Pulmonary Vascular Diseases Consortium: United against a Global Health Challenge. Pulm. Circ. 2024, 14, e70003. [Google Scholar] [CrossRef]
- Butrous, G.; Mathie, A. Infection in Pulmonary Vascular Diseases: Would Another Consortium Really Be the Way to Go? Glob. Cardiol. Sci. Pract. 2019, 2019, 1. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic Definitions and Updated Clinical Classification of Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Melot, C.; Naeije, R. Pulmonary Vascular Diseases. Compr. Physiol. 2011, 1, 593–619. [Google Scholar]
- Hatano, S.; Strasser, T.; World Health Organization. Primary Pulmonary Hypertension: Report on a WHO Meeting, Geneva, 15–17 October 1973; World Health Organization: Geneva, Switzerland, 1975; ISBN 92-4-156044-4. [Google Scholar]
- Kovacs, G.; Maier, R.; Aberer, E.; Brodmann, M.; Scheidl, S.; Tröster, N.; Hesse, C.; Salmhofer, W.; Graninger, W.; Gruenig, E. Borderline Pulmonary Arterial Pressure Is Associated with Decreased Exercise Capacity in Scleroderma. Am. J. Respir. Crit. Care Med. 2009, 180, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Emmons-Bell, S.; Johnson, C.; Boon-Dooley, A.; Corris, P.A.; Leary, P.J.; Rich, S.; Yacoub, M.; Roth, G.A. Prevalence, Incidence, and Survival of Pulmonary Arterial Hypertension: A Systematic Review for the Global Burden of Disease 2020 Study. Pulm. Circ. 2022, 12, e12020. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, L.; Droppa, M. Pulmonary Hypertension and COVID-19. Hamostaseologie 2022, 42, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Potus, F.; Mai, V.; Lebret, M.; Malenfant, S.; Breton-Gagnon, E.; Lajoie, A.C.; Boucherat, O.; Bonnet, S.; Provencher, S. Novel Insights on the Pulmonary Vascular Consequences of COVID-19. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L277–L288. [Google Scholar] [CrossRef]
- Pagnesi, M.; Baldetti, L.; Beneduce, A.; Calvo, F.; Gramegna, M.; Pazzanese, V.; Ingallina, G.; Napolano, A.; Finazzi, R.; Ruggeri, A. Pulmonary Hypertension and Right Ventricular Involvement in Hospitalised Patients with COVID-19. Heart 2020, 106, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Aktay-Cetin, Ö.; Craddock, V.; Morales-Cano, D.; Kosanovic, D.; Cogolludo, A.; Perez-Vizcaino, F.; Avdeev, S.; Kumar, A.; Ram, A.K. Potential Long-Term Effects of SARS-CoV-2 Infection on the Pulmonary Vasculature: Multilayered Cross-Talks in the Setting of Coinfections and Comorbidities. PLoS Pathog. 2023, 19, e1011063. [Google Scholar] [CrossRef] [PubMed]
- Musuuza, J.S.; Watson, L.; Parmasad, V.; Putman-Buehler, N.; Christensen, L.; Safdar, N. Prevalence and Outcomes of Co-Infection and Superinfection with SARS-CoV-2 and Other Pathogens: A Systematic Review and Meta-Analysis. PLoS ONE 2021, 16, e0251170. [Google Scholar] [CrossRef]
- Egom, E.À.; Shiwani, H.A.; Nouthe, B. From Acute SARS-CoV-2 Infection to Pulmonary Hypertension. Front. Physiol. 2022, 13, 1023758. [Google Scholar]
- Taha, H.A.; Elshafey, B.I.; Abdullah, T.M.; Salem, H.A. Study of Pulmonary Hypertension in Post-COVID-19 Patients by Transthoracic Echocardiography. Egypt. J. Bronchol. 2023, 17, 27. [Google Scholar] [CrossRef]
- Oktaviono, Y.H.; Mulia, E.P.B.; Luke, K.; Nugraha, D.; Maghfirah, I.; Subagjo, A. Right Ventricular Dysfunction and Pulmonary Hypertension in COVID-19: A Meta-Analysis of Prevalence and Its Association with Clinical Outcome. Arch. Med. Sci. AMS 2022, 18, 1169. [Google Scholar] [CrossRef] [PubMed]
- Butrous, G. Human Immunodeficiency Virus–Associated Pulmonary Arterial Hypertension: Considerations for Pulmonary Vascular Diseases in the Developing World. Circulation 2015, 131, 1361–1370. [Google Scholar] [CrossRef]
- Medrano-Garcia, S.; Morales-Cano, D.; Barreira, B.; Vera-Zambrano, A.; Kumar, R.; Kosanovic, D.; Schermuly, R.T.; Graham, B.B.; Perez-Vizcaino, F.; Mathie, A.; et al. HIV and Schistosoma Co-Exposure Leads to Exacerbated Pulmonary Endothelial Remodeling and Dysfunction Associated with Altered Cytokine Landscape. Cells 2022, 11, 2414. [Google Scholar] [CrossRef] [PubMed]
- Butrous, G. The Global Challenge of Pulmonary Vascular Diseases and Its Forgotten Impact in the Developing World. Adv. Pulm. Hypertens. 2012, 11, 117–118. [Google Scholar] [CrossRef]
- Butrous, G. Pulmonary Vascular Diseases Secondary to Schistosomiasis. Adv. Pulm. Hypertens. 2017, 15, 144–148. [Google Scholar] [CrossRef]
- Kolosionek, E.; Crosby, A.; Harhay, M.O.; Morrell, N.; Butrous, G. Pulmonary Vascular Disease Associated with Schistosomiasis. Expert. Rev. Anti-Infect. Ther. 2010, 8, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- WHO. Status of Schistosomiasis Endemic Countries: 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Xue, Q.; Deng, Y.; Liu, Y.; Wang, Y.; Hu, W.; Huang, Y.; Yang, K. A Retrospective Analysis of Schistosomiasis Related Literature from 2011-2020: Focusing on the next Decade. Acta Trop. 2023, 238, 106750. [Google Scholar] [CrossRef] [PubMed]
- Rohun, J.; Dorniak, K.; Faran, A.; Kochańska, A.; Zacharek, D.; Daniłowicz-Szymanowicz, L. Long COVID-19 Myocarditis and Various Heart Failure Presentations: A Case Series. J. Cardiovasc. Dev. Dis. 2022, 9, 427. [Google Scholar] [CrossRef]
- Obeyesekere, I.; Peiris, D. Pulmonary Hypertension and Filariasis. Br. Heart J. 1974, 36, 676–681. [Google Scholar] [CrossRef]
- Walloopillai, N. Primary Pulmonary Hypertension, an Unexplained Epidemic in Sri Lanka. Pathobiology 1975, 43, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Wolday, D.; Gebrecherkos, T.; Arefaine, Z.G.; Kiros, Y.K.; Gebreegzabher, A.; Tasew, G.; Abdulkader, M.; Abraha, H.E.; Desta, A.A.; Hailu, A. Effect of Co-Infection with Intestinal Parasites on COVID-19 Severity: A Prospective Observational Cohort Study. EClinicalMedicine 2021, 39, 101054. [Google Scholar] [CrossRef]
- Gebrecherkos, T.; Gessesse, Z.; Kebede, Y.; Gebreegzabher, A.; Tasew, G.; Abdulkader, M.; Ebuy, H.; Desta, A.; Hailu, A.; Harris, V. Effect of Co-Infection with Parasites on Severity of COVID-19. medRxiv 2021, 2021, 101054. [Google Scholar]
- Lai, K.; McFadzean, A.; Yeung, R. Microembolic Pulmonary Hypertension in Pyogenic Cholangitis. Br. Med. J. 1968, 1, 22. [Google Scholar] [CrossRef]
- Ross, A.G.; Sleigh, A.C.; Li, Y.; Davis, G.M.; Williams, G.M.; Jiang, Z.; Feng, Z.; McManus, D.P. Schistosomiasis in the People’s Republic of China: Prospects and Challenges for the 21st Century. Clin. Microbiol. Rev. 2001, 14, 270–295. [Google Scholar] [CrossRef]
- Halawa, S.; Pullamsetti, S.S.; Bangham, C.R.; Stenmark, K.R.; Dorfmüller, P.; Frid, M.G.; Butrous, G.; Morrell, N.W.; de Jesus Perez, V.A.; Stuart, D.I. Potential Long-Term Effects of SARS-CoV-2 Infection on the Pulmonary Vasculature: A Global Perspective. Nat. Rev. Cardiol. 2022, 19, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.K.; Nyasulu, P.S.; Iqbal, A.A.; Hamdan Gul, M.; Ferreira, E.V.; Leclair, J.W.; Htun, Z.M.; Howard, L.S.; Mocumbi, A.O.; Bryant, A.J. Cardiopulmonary Disease as Sequelae of Long-Term COVID-19: Current Perspectives and Challenges. Front. Med. 2022, 9, 1041236. [Google Scholar] [CrossRef] [PubMed]
- Nijkeuter, M.; Hovens, M.M.; Davidson, B.L.; Huisman, M.V. Resolution of Thromboemboli in Patients with Acute Pulmonary Embolism: A Systematic Review. Chest 2006, 129, 192–197. [Google Scholar] [CrossRef]
- Pullamsetti, S.; Savai, R.; Janssen, W.; Dahal, B.; Seeger, W.; Grimminger, F.; Ghofrani, H.; Weissmann, N.; Schermuly, R. Inflammation, Immunological Reaction and Role of Infection in Pulmonary Hypertension. Clin. Microbiol. Infect. 2011, 17, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J. Pulmonary Arterial Hypertension. Proc. Am. Thorac. Soc. 2006, 3, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial Dysfunction in Pulmonary Hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef]
- Hassoun, P.M.; Mouthon, L.; Barberà, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; Jones, P.L.; Maitland, M.L.; Michelakis, E.D.; Morrell, N.W.; et al. Inflammation, Growth Factors, and Pulmonary Vascular Remodeling. J. Am. Coll. Cardiol. 2009, 54, S10–S19. [Google Scholar] [CrossRef]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef] [PubMed]
- Ogo, T.; Chowdhury, H.; Yang, J.; Long, L.; Li, X.; Torres Cleuren, Y.N.; Morrell, N.W.; Schermuly, R.T.; Trembath, R.C.; Nasim, M.T. Inhibition of Overactive Transforming Growth Factor–β Signaling by Prostacyclin Analogs in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 48, 733–741. [Google Scholar] [CrossRef]
- Burel-Vandenbos, F.; Cardot-Leccia, N.; Passeron, T. Apoptosis and Pericyte Loss in Alveolar Capillaries in COVID-19 Infection: Choice of Markers Matters. Author’s Reply. Intensive Care Med. 2020, 46, 1967–1968. [Google Scholar] [CrossRef]
- Oyeyemi, O.; Okunlola, O.; Adebayo, A. Assessment of Schistosomiasis Endemicity and Preventive Treatment on Coronavirus Disease 2019 Outcomes in Africa. New Microbes New Infect. 2020, 38, 100821. [Google Scholar] [CrossRef]
- Maizels, R.M.; McSorley, H.J. Regulation of the Host Immune System by Helminth Parasites. J. Allergy Clin. Immunol. 2016, 138, 666–675. [Google Scholar] [CrossRef]
- Ferrari, T.C.A.; Albricker, A.C.L.; Gonçalves, I.M.; Freire, C.M.V. Schistosome-Associated Pulmonary Arterial Hypertension: A Review Emphasizing Pathogenesis. Front. Cardiovasc. Med. 2021, 8, 724254. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mickael, C.; Chabon, J.; Gebreab, L.; Rutebemberwa, A.; Garcia, A.R.; Koyanagi, D.E.; Sanders, L.; Gandjeva, A.; Kearns, M.T. The Causal Role of IL-4 and IL-13 in Schistosoma mansoni Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Butrous, G.; Ghofrani, H.A.; Grimminger, F. Pulmonary Vascular Disease in the Developing World. Circulation 2008, 118, 1758–1766. [Google Scholar] [CrossRef]
- Chiu, B.-C.; Freeman, C.M.; Stolberg, V.R.; Komuniecki, E.; Lincoln, P.M.; Kunkel, S.L.; Chensue, S.W. Cytokine–Chemokine Networks in Experimental Mycobacterial and Schistosomal Pulmonary Granuloma Formation. Am. J. Respir. Cell Mol. Biol. 2003, 29, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Boros, D.L.; Whitfield, J.R. Enhanced Th1 and Dampened Th2 Responses Synergize to Inhibit Acute Granulomatous and Fibrotic Responses in Murine Schistosomiasis Mansoni. Infect. Immun. 1999, 67, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Noor, R. How Do the Severe Acute Respiratory Coronavirus 2 (SARS-CoV-2) and Its Variants Escape the Host Protective Immunity and Mediate Pathogenesis? Bull. Natl. Res. Cent. 2022, 46, 255. [Google Scholar] [CrossRef] [PubMed]
- D Avila-Mesquita, C.; Couto, A.E.S.; Campos, L.C.B.; Vasconcelos, T.F.; Michelon-Barbosa, J.; Corsi, C.A.C.; Mestriner, F.; Petroski-Moraes, B.C.; Garbellini-Diab, M.J.; Couto, D.M.S.; et al. MMP-2 and MMP-9 Levels in Plasma Are Altered and Associated with Mortality in COVID-19 Patients. Biomed. Pharmacother. 2021, 142, 112067. [Google Scholar] [CrossRef] [PubMed]
- Laveaux, S.; Vandecasteele, S.; Van De Moortele, K. Chronic Schistosomiasis Presenting with Migrating Pulmonary Manifestation after Recent COVID-19 Infection: HRCT Findings. J. Belg. Soc. Radiol. 2022, 106, 21. [Google Scholar] [CrossRef]
- Turner, J.D.; Jackson, J.A.; Faulkner, H.; Behnke, J.; Else, K.J.; Kamgno, J.; Boussinesq, M.; Bradley, J.E. Intensity of Intestinal Infection with Multiple Worm Species Is Related to Regulatory Cytokine Output and Immune Hyporesponsiveness. J. Infect. Dis. 2008, 197, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Babu, S.; Nutman, T.B. Immunology of Lymphatic Filariasis. Parasite Immunol. 2014, 36, 338–346. [Google Scholar] [CrossRef]
- Mahajan, G.; Barjatya, H.; Bhakar, B.; Gothwal, S.K.; Jangir, T. To Estimate Prevalence of Pulmonary Arterial Hypertension in HIV Patients and Its Association with CD4 Cell Count. Clin. Epidemiol. Glob. Health 2024, 25, 101479. [Google Scholar] [CrossRef]
- UNAIDS. Global HIV Statistics; Joint United Nations Programme on HIV/AIDS: Geneva, Switzerland, 2023. [Google Scholar]
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.-C.; Gibbs, J.S.R. A Global View of Pulmonary Hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef]
- Almodovar, S.; Cicalini, S.; Petrosillo, N.; Flores, S.C. Pulmonary Hypertension Associated with HIV Infection: Pulmonary Vascular Disease: The Global Perspective. Chest 2010, 137, 6S–12S. [Google Scholar] [CrossRef]
- Agarwal, S.; Sharma, H.; Chen, L.; Dhillon, N.K. NADPH Oxidase-Mediated Endothelial Injury in HIV- and Opioid-Induced Pulmonary Arterial Hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L1097–L1108. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.M.; Walp, E.R.; Elms, S.C.; Raynor, R.; Mitchell, P.O.; Guidot, D.M.; Sutliff, R.L. Human Immunodeficiency Virus-1 Transgene Expression Increases Pulmonary Vascular Resistance and Exacerbates Hypoxia-Induced Pulmonary Hypertension Development. Pulm. Circ. 2013, 3, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Nuche, J.; Pérez-Olivares, C.; de la Cal, T.S.; López-Guarch, C.J.; Ynsaurriaga, F.A.; Subías, P.E. Clinical Course of COVID-19 in Pulmonary Arterial Hypertension Patients. Rev. Esp. De Cardiol. Engl. Ed. 2020, 73, 775. [Google Scholar] [CrossRef]
- Danwang, C.; Noubiap, J.J.; Robert, A.; Yombi, J.C. Outcomes of Patients with HIV and COVID-19 Co-Infection: A Systematic Review and Meta-Analysis. AIDS Res. Ther. 2022, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.J.; Khan, I.A.; Mehta, R.N.; Sepkowitz, D.A. HIV-Related Pulmonary Hypertension: Analytic Review of 131 Cases. Chest 2000, 118, 1133–1141. [Google Scholar] [CrossRef]
- Suresh, S.J.; Suzuki, Y.J. SARS-CoV-2 Spike Protein and Lung Vascular Cells. J. Respir. 2020, 1, 40–48. [Google Scholar] [CrossRef]
- Dorfmüller, P.; Perros, F.; Balabanian, K.; Humbert, M. Inflammation in Pulmonary Arterial Hypertension. Eur. Respir. J. 2003, 22, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Butrous, G. Human Immunodeficiency Viruses and Their Effect on the Pulmonary Vascular Bed. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 321, L1062–L1066. [Google Scholar] [CrossRef] [PubMed]
- Cool, C.D.; Rai, P.R.; Yeager, M.E.; Hernandez-Saavedra, D.; Serls, A.E.; Bull, T.M.; Geraci, M.W.; Brown, K.K.; Routes, J.M.; Tuder, R.M. Expression of Human Herpesvirus 8 in Primary Pulmonary Hypertension. N. Engl. J. Med. 2003, 349, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.M.; Lederer, D.J.; Borczuk, A.C.; Kawut, S.M. Pulmonary Hypertension in Idiopathic Pulmonary Fibrosis. Chest 2007, 132, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, A.; Longhurst, H.J.; Paxton, J.K.; Scotton, C.J. The Role of Herpes Viruses in Pulmonary Fibrosis. Front. Med. 2021, 8, 704222. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Kipar, A.; Lunardi, F.; Balestro, E.; Perissinotto, E.; Rossi, E.; Nannini, N.; Marulli, G.; Stewart, J.P.; Rea, F. Herpes Virus Infection Is Associated with Vascular Remodeling and Pulmonary Hypertension in Idiopathic Pulmonary Fibrosis. PLoS ONE 2013, 8, e55715. [Google Scholar] [CrossRef] [PubMed]
- Zubchenko, S.; Kril, I.; Nadizhko, O.; Matsyura, O.; Chopyak, V. Herpesvirus Infections and Post-COVID-19 Manifestations: A Pilot Observational Study. Rheumatol. Int. 2022, 42, 1523–1530. [Google Scholar] [CrossRef]
- Mohamed, A.; Gidda, H.; Zavoshi, S.; Mahmood, R. A Case of Left Ventricular Thrombus and Herpetic Esophagitis in an Immunocompetent Patient With COVID-19. Cureus 2023, 15, e33640. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2025; World Health Organization: Geneva, Switzerland, 2025. [Google Scholar]
- Bhattacharyya, P.; Saha, D.; Bhattacherjee, P.D.; Das, S.K.; Bhattacharyya, P.P.; Dey, R. Tuberculosis Associated Pulmonary Hypertension: The Revelation of a Clinical Observation. Lung India Off. Organ. Indian. Chest Soc. 2016, 33, 135. [Google Scholar] [CrossRef]
- Louw, E.; Baines, N.; Maarman, G.; Osman, M.; Sigwadhi, L.; Irusen, E.; Koegelenberg, C.; Doubell, A.; Nathan, S.; Channick, R. The Prevalence of Pulmonary Hypertension after Successful Tuberculosis Treatment in a Community Sample of Adult Patients. Pulm. Circ. 2023, 13, e12184. [Google Scholar] [CrossRef]
- Galie, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.-L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S. Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), Endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar]
- Tamuzi, J.L.; Lulendo, G.; Mbuesse, P.; Nyasulu, P.S. The Incidence and Mortality of COVID-19 Related TB Disease in Sub-Saharan Africa: A Systematic Review and Meta-Analysis. medRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Ahmed, A.E.H.; Ibrahim, A.S.; Elshafie, S.M. Pulmonary Hypertension in Patients with Treated Pulmonary Tuberculosis: Analysis of 14 Consecutive Cases. Clin. Med. Insights: Circ. Respir. Pulm. Med. 2011, 5, CCRPM-S6437. [Google Scholar] [CrossRef]
- Jo, Y.S.; Park, J.-H.; Lee, J.K.; Heo, E.Y.; Chung, H.S.; Kim, D.K. Risk Factors for Pulmonary Arterial Hypertension in Patients with Tuberculosis-Destroyed Lungs and Their Clinical Characteristics Compared with Patients with Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 2433–2443. [Google Scholar] [CrossRef] [PubMed]
- Kayongo, A.; Nyiro, B.; Siddharthan, T.; Kirenga, B.; Checkley, W.; Lutaakome Joloba, M.; Ellner, J.; Salgame, P. Mechanisms of Lung Damage in Tuberculosis: Implications for Chronic Obstructive Pulmonary Disease. Front. Cell Infect. Microbiol. 2023, 13, 1146571. [Google Scholar] [CrossRef]
- Ernst, J.D. The Immunological Life Cycle of Tuberculosis. Nat. Rev. Immunol. 2012, 12, 581–591. [Google Scholar] [CrossRef]
- Russell, D.G.; Cardona, P.-J.; Kim, M.-J.; Allain, S.; Altare, F. Foamy Macrophages and the Progression of the Human Tuberculosis Granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S. Pathogenesis of Cor Pulmonale in Pulmonary Tuberculosis. Ind. J. Tuberc. 1986, 33, 167–170. [Google Scholar]
- Zouaki, I.; Chahbi, Z.; Raiteb, M.; Zyani, M. COVID-19 and Pulmonary Tuberculosis Coinfection in a Moroccan Patient with Pulmonary Embolism: A Case Report and Literature Review. Case Rep. Infect. Dis. 2022, 2022, 1522876. [Google Scholar] [CrossRef] [PubMed]
- Parolina, L.; Pshenichnaya, N.; Vasilyeva, I.; Lizinfed, I.; Urushadze, N.; Guseva, V.; Otpushchennikova, O.; Dyachenko, O.; Kharitonov, P. Clinical Characteristics of COVID-19 in Patients with Tuberculosis and Factors Associated with the Disease Severity. Int. J. Infect. Dis. 2022, 124, S82–S89. [Google Scholar] [CrossRef]
- He, F.; Xia, X.; Nie, D.; Yang, H.; Jiang, Y.; Huo, X.; Guo, F.; Fang, B.; Hu, B.; Jiang, H.; et al. Respiratory Bacterial Pathogen Spectrum among COVID-19 Infected and Non-COVID-19 Virus Infected Pneumonia Patients. Diagn. Microbiol. Infect. Dis. 2020, 98, 115199. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, Y.; Liu, X.; Fu, Y.; Zhang, J.; Zhang, Y.; Zhang, L.; Wei, X.; Yang, L. Systematic Review and Meta-Analysis of Tuberculosis and COVID-19 Co-Infection: Prevalence, Fatality, and Treatment Considerations. PLoS Negl. Trop. Dis. 2024, 18, e0012136. [Google Scholar] [CrossRef]
- Berger, J.T.; Carcillo, J.A.; Shanley, T.P.; Wessel, D.L.; Clark, A.; Holubkov, R.; Meert, K.L.; Newth, C.J.L.; Berg, R.A.; Heidemann, S.; et al. Critical Pertussis Illness in Children: A Multicenter Prospective Cohort Study. Pediatr. Crit. Care Med. 2013, 14, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Jeican, I.I.; Inișca, P.; Gheban, D.; Tăbăran, F.; Aluaș, M.; Trombitas, V.; Cristea, V.; Crivii, C.; Junie, L.M.; Albu, S. COVID-19 and Pneumocystis Jirovecii Pulmonary Coinfection—The First Case Confirmed through Autopsy. Medicina 2021, 57, 302. [Google Scholar] [CrossRef] [PubMed]
- Saibaba, J.; Selvaraj, J.; Viswanathan, S.; Pillai, V.; Saibaba, J., Jr. Scrub Typhus and COVID-19 Coinfection Unmasking Antiphospholipid Antibody Syndrome. Cureus 2022, 14, e25008. [Google Scholar] [CrossRef] [PubMed]
- Udwadia, Z.F.; Vora, A.; Tripathi, A.R.; Malu, K.N.; Lange, C.; Raju, R.S. COVID-19-Tuberculosis Interactions: When Dark Forces Collide. Indian J. Tuberc. 2020, 67, S155–S162. [Google Scholar] [CrossRef]
- Tamuzi, J.L.; Ayele, B.T.; Shumba, C.S.; Adetokunboh, O.O.; Uwimana-Nicol, J.; Haile, Z.T.; Inugu, J.; Nyasulu, P.S. Implications of COVID-19 in High Burden Countries for HIV/TB: A Systematic Review of Evidence. BMC Infect. Dis. 2020, 20, 744. [Google Scholar] [CrossRef]
- Tsenova, L.; Singhal, A. Effects of Host-Directed Therapies on the Pathology of Tuberculosis. J. Pathol. 2020, 250, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, V.; Gualano, G.; Musso, M.; Libertone, R.; Nisii, C.; Ianniello, S.; Mosti, S.; Mastrobattista, A.; Cerva, C.; Bevilacqua, N. Increased Association of Pulmonary Thromboembolism and Tuberculosis during COVID-19 Pandemic: Data from an Italian Infectious Disease Referral Hospital. Antibiotics 2022, 11, 398. [Google Scholar] [CrossRef]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH Interim Guidance on Recognition and Management of Coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef]
- Paddock, C.D.; Sanden, G.N.; Cherry, J.D.; Gal, A.A.; Langston, C.; Tatti, K.M.; Wu, K.-H.; Goldsmith, C.S.; Greer, P.W.; Montague, J.L.; et al. Pathology and Pathogenesis of Fatal Bordetella Pertussis Infection in Infants. Clin. Infect. Dis. 2008, 47, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.H.T.; Duclos, P.; Nelson, E.A.S.; Hutubessy, R.C.W. An Update of the Global Burden of Pertussis in Children Younger than 5 Years: A Modelling Study. Lancet Infect. Dis. 2017, 17, 974–980. [Google Scholar] [CrossRef]
- Shi, T.; Wang, L.; Du, S.; Fan, H.; Yu, M.; Ding, T.; Xu, X.; Zhang, D.; Huang, L.; Lu, G. Mortality Risk Factors among Hospitalized Children with Severe Pertussis. BMC Infect. Dis. 2021, 21, 1057. [Google Scholar] [CrossRef] [PubMed]
- Flikweert, A.W.; Grootenboers, M.J.; Yick, D.C.; du Mee, A.W.; van der Meer, N.J.; Rettig, T.C.; Kant, M.K. Late Histopathologic Characteristics of Critically Ill COVID-19 Patients: Different Phenotypes without Evidence of Invasive Aspergillosis, a Case Series. J. Crit. Care 2020, 59, 149–155. [Google Scholar] [CrossRef]
- van Arkel, A.L.; Rijpstra, T.A.; Belderbos, H.N.; Van Wijngaarden, P.; Verweij, P.E.; Bentvelsen, R.G. COVID-19–Associated Pulmonary Aspergillosis. Am. J. Respir. Crit. Care Med. 2020, 202, 132–135. [Google Scholar] [CrossRef]
- Kamath, A.; Mishra, G.; Munje, R.; Atram, J. Post-COVID Pulmonary Aspergillosis with Pulmonary Thromboembolism and Pulmonary Artery Hypertension Unmasking Prediabetes: A Case Report. Vidarbha J. Intern. Med. 2023, 33, 42–45. [Google Scholar] [CrossRef]
- Lamoth, F.; Glampedakis, E.; Boillat-Blanco, N.; Oddo, M.; Pagani, J.-L. Incidence of Invasive Pulmonary Aspergillosis among Critically Ill COVID-19 Patients. Clin. Microbiol. Infect. 2020, 26, 1706–1708. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, P.; Mallat, J.; Arumadura, C.; Vangrunderbeek, N.; Dupre, C.; Pauquet, P.; Orfi, A.; Granier, M.; Lemyze, M. Risk Factors for Invasive Pulmonary Aspergillosis in Critically Ill Patients with Coronavirus Disease 2019-Induced Acute Respiratory Distress Syndrome. Crit. Care Explor. 2020, 2, e0244. [Google Scholar] [CrossRef]
- Chit Yee, D.; Aung, H.K.K.; Mg Mg, B.; Htun, W.P.P.; Janurian, N.; Pyae Phyo, A.; Bancone, G.; Watthanaworawit, W.; Proux, S.; Nosten, F. Case Report: A Case Report of Multiple Co-Infections (Melioidosis, Paragonimiasis, COVID-19 and Tuberculosis) in a Patient with Diabetes Mellitus and Thalassemia-Trait in Myanmar. Wellcome Open Res. 2022, 7, F1000. [Google Scholar] [CrossRef]
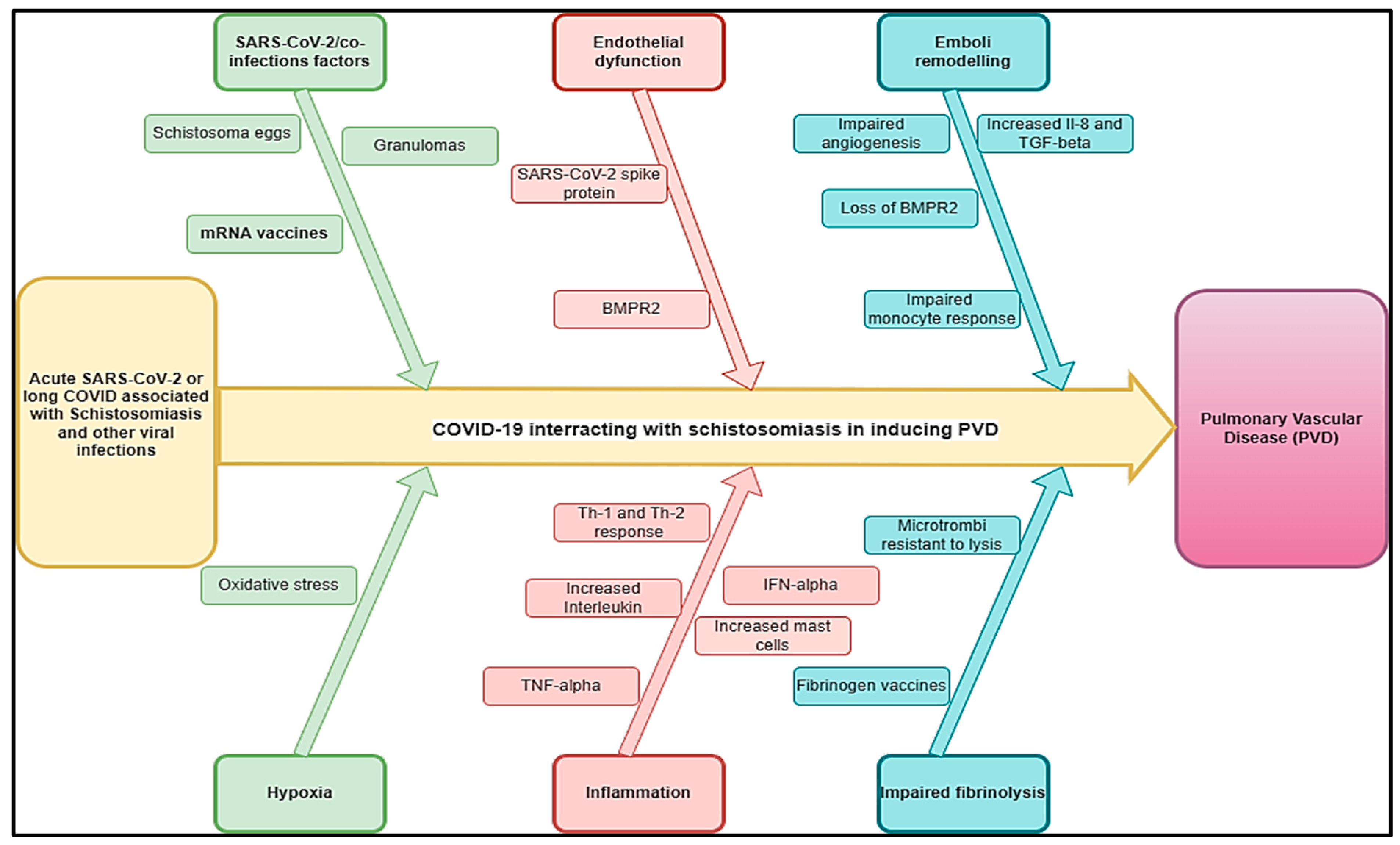
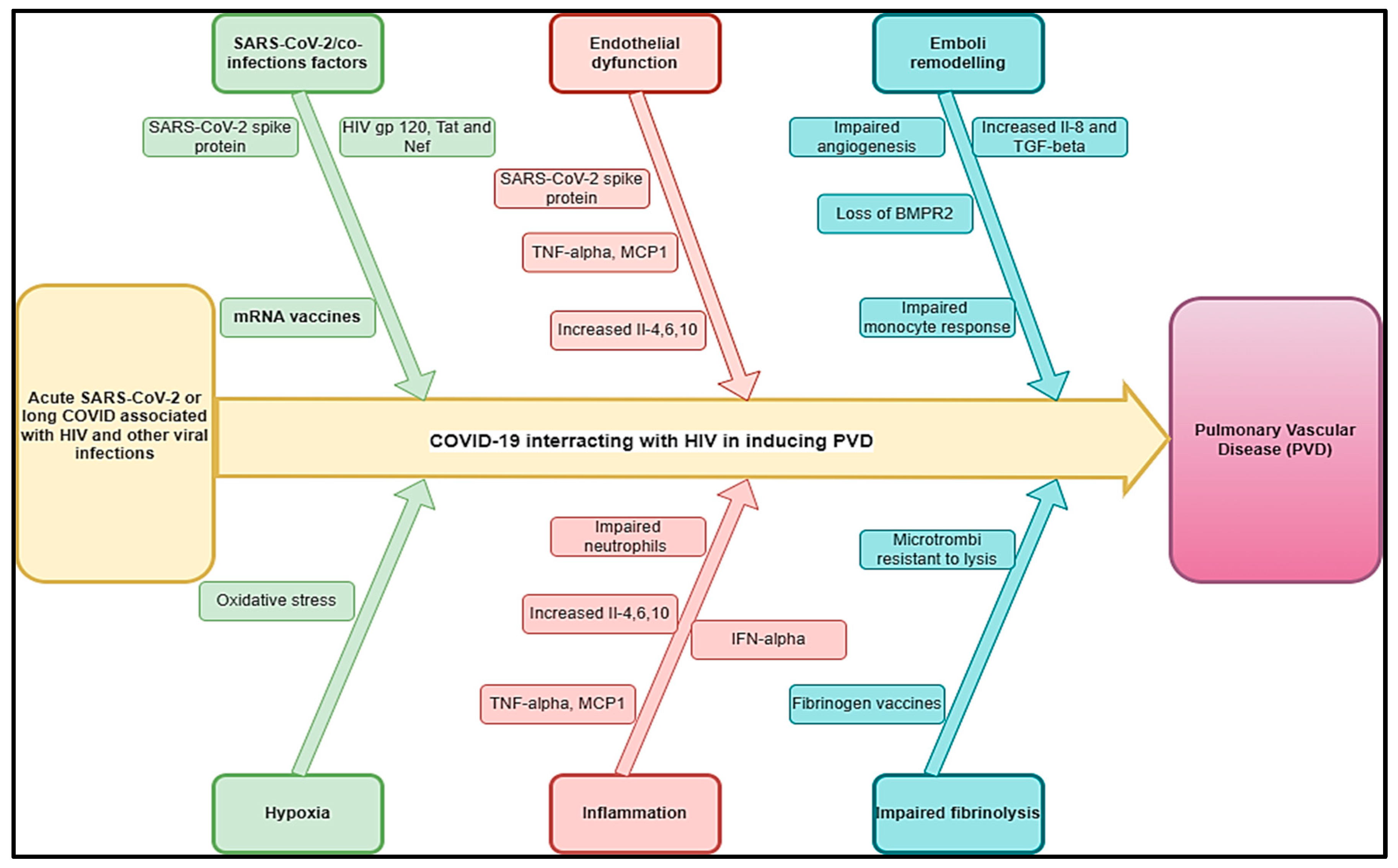

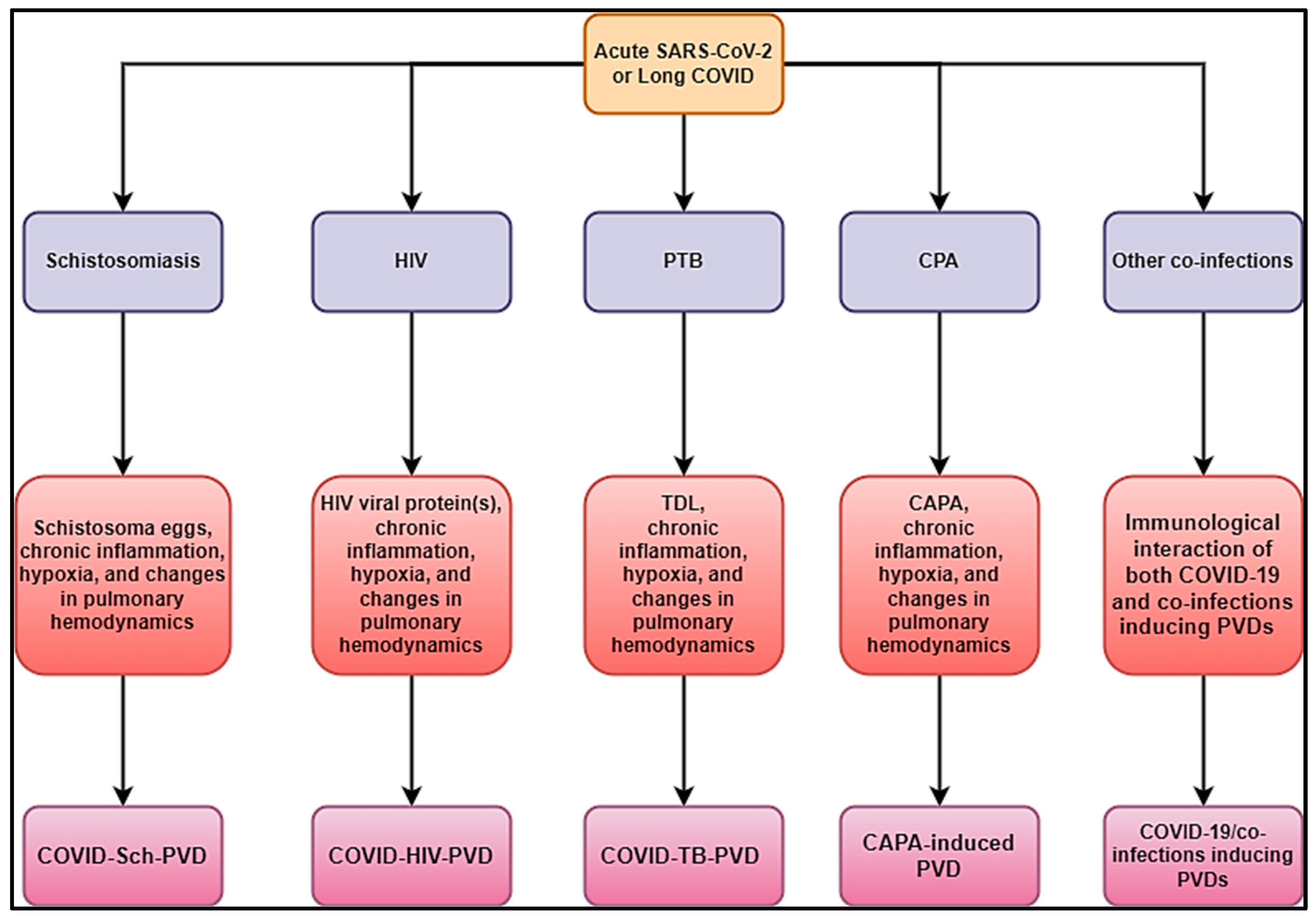


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research Gaps | Plausible Mechanisms Inducing PVDs | Hypothesis | Future Perspectives |
|---|---|---|---|
| COVID-19 on schistosomiasis-inducing PVD (COVID-Sch-PVD) cases and mechanisms | Schistosoma eggs, chronic inflammation and immunological hyperactivation, chronic hypoxia, and changes in pulmonary hemodynamics, such as endothelial dysfunction, vascular leak, and thrombotic microangiopathy, are all associated with acute or long-term COVID. | How severe could COVID-Sch-PVD be compared to Sch-PVD? How could the clinical picture of COVID-Sch-PVD found in people with a history of Sch-PVD or COVID-Sch-PVD be found in the long COVID phase? What could be the most plausible pathophysiological pathways of COVID-Sch-PVD be? | Active clinical case search and subsequent investigation in high-burden COVID-19 and schistosomiasis settings in PVD cases. More experimental studies using small animal, large animal, and in vitro models. |
| COVID-19 associated with HIV inducing PVD (COVID-HIV-PVD) cases and mechanisms | HIV-viral protein(s), chronic inflammation and immune hyperactivation, chronic hypoxia, and alterations in pulmonary hemodynamics, including endothelial dysfunction, vascular leak, and thrombotic micro-angiopathy due to acute or long COVID. | How severe could COVID-HIV-PVD be compared to HIV-PVD? How could the clinical picture of COVID-HIV-PVD found in people with a history of HIV-PVD or COVID-HIV-PVD be found in the long COVID phase? What could be the most plausible pathophysiological pathways of COVID-HIV-PVD be? | Active clinical case search and subsequent investigation in high-burden COVID-19 and HIV settings in PVD cases. More experimental studies using small animal, large animal, and in vitro models. |
| COVID-19 on TB inducing PVDs (COVID-TB-PVD cases and mechanisms | TB-destroyed lung (TDL), chronic inflammation and immunological hyperactivation, chronic hypoxia, and changes in pulmonary hemodynamics, such as endothelial dysfunction, vascular leak, and thrombotic microangiopathy, as a result of acute or prolonged COVID. | How severe is COVID-TB-PVD in comparison to TB-PVD? How might the clinical picture of COVID-TB-PVD be found in people with a history of TB-PVD, or could it be found throughout the extended COVID phase? What are the most likely pathophysiological routes of COVID-TB-PVD? | Active clinical case search and subsequent investigation in high-burden COVID-19 and TB settings in PVD cases. More experimental studies using small animal, large animal, and in vitro models. |
| CAPA-induced PVDs cases and mechanisms | Chronic pulmonary aspergillosis (CPA), chronic inflammation and immune hyperactivation, chronic hypoxia, and alterations in pulmonary hemodynamics, including endothelial dysfunction, vascular leak, and thrombotic micro-angiopathy due to acute or long COVID. | How severe is CAPA-induc ing PVD in comparison to CPA-inducing PVD? What are the most likely pathophysiological routes of CAPA-inducing PVD? | Active clinical case search and subsequent study in high-burden filariasis and COVID-19 situations in PVD cases. More experimental research with small and big animal models, as well as in vitro models. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nyasulu, P.S.; Tamuzi, J.L.; Oliveira, R.K.F.; Oliveira, S.D.; Petrosillo, N.; de Jesus Perez, V.; Dhillon, N.; Butrous, G. COVID-19 and Parasitic Co-Infection: A Hypothetical Link to Pulmonary Vascular Disease. Infect. Dis. Rep. 2025, 17, 19. https://doi.org/10.3390/idr17020019
Nyasulu PS, Tamuzi JL, Oliveira RKF, Oliveira SD, Petrosillo N, de Jesus Perez V, Dhillon N, Butrous G. COVID-19 and Parasitic Co-Infection: A Hypothetical Link to Pulmonary Vascular Disease. Infectious Disease Reports. 2025; 17(2):19. https://doi.org/10.3390/idr17020019
Chicago/Turabian StyleNyasulu, Peter S., Jacques L. Tamuzi, Rudolf K. F. Oliveira, Suellen D. Oliveira, Nicola Petrosillo, Vinicio de Jesus Perez, Navneet Dhillon, and Ghazwan Butrous. 2025. "COVID-19 and Parasitic Co-Infection: A Hypothetical Link to Pulmonary Vascular Disease" Infectious Disease Reports 17, no. 2: 19. https://doi.org/10.3390/idr17020019
APA StyleNyasulu, P. S., Tamuzi, J. L., Oliveira, R. K. F., Oliveira, S. D., Petrosillo, N., de Jesus Perez, V., Dhillon, N., & Butrous, G. (2025). COVID-19 and Parasitic Co-Infection: A Hypothetical Link to Pulmonary Vascular Disease. Infectious Disease Reports, 17(2), 19. https://doi.org/10.3390/idr17020019