

Khosta: A Genetic and Structural Point of View of the Forgotten Virus

 ,
,  , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
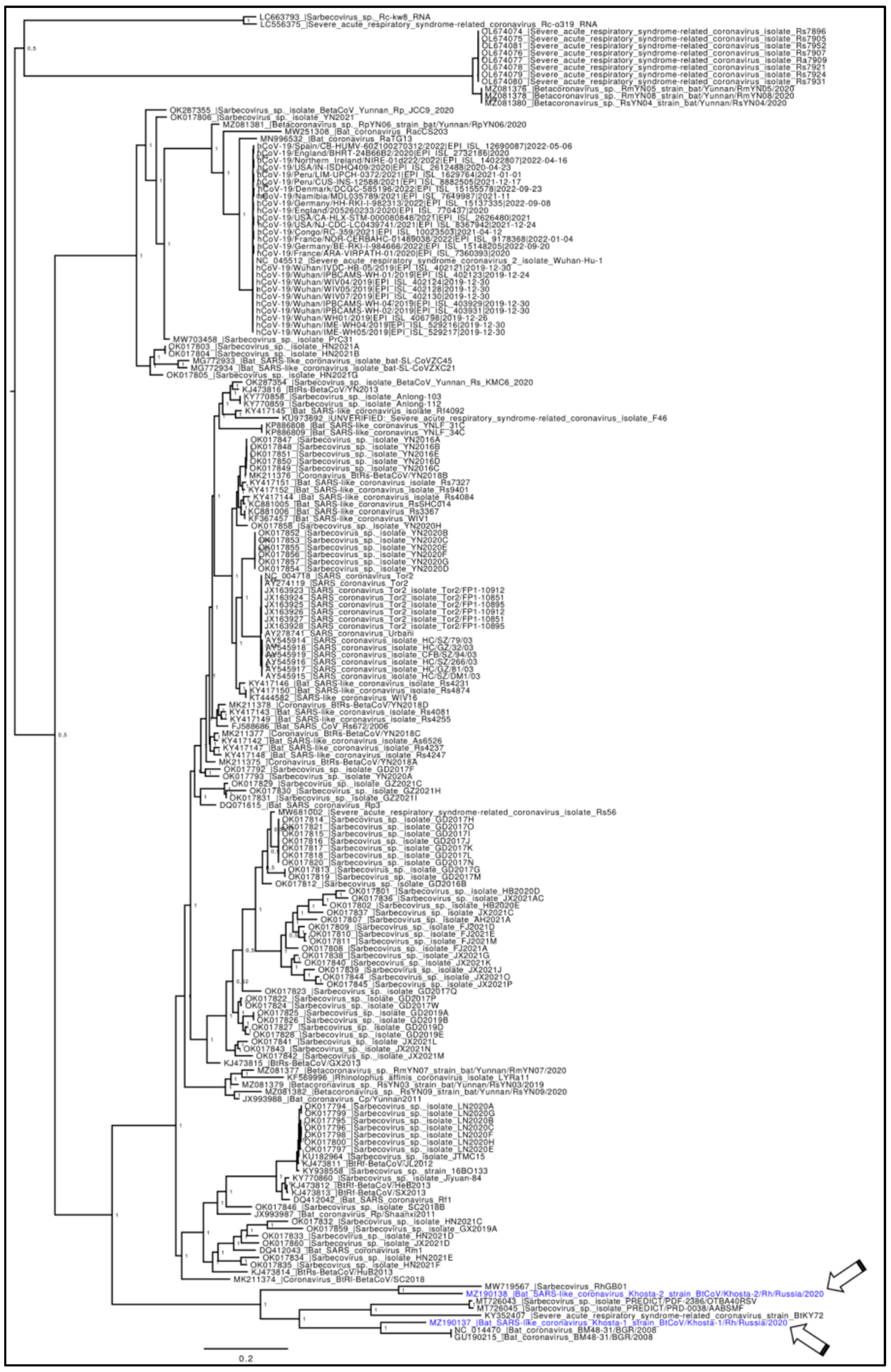
2.1. Phylogenetic Analyses
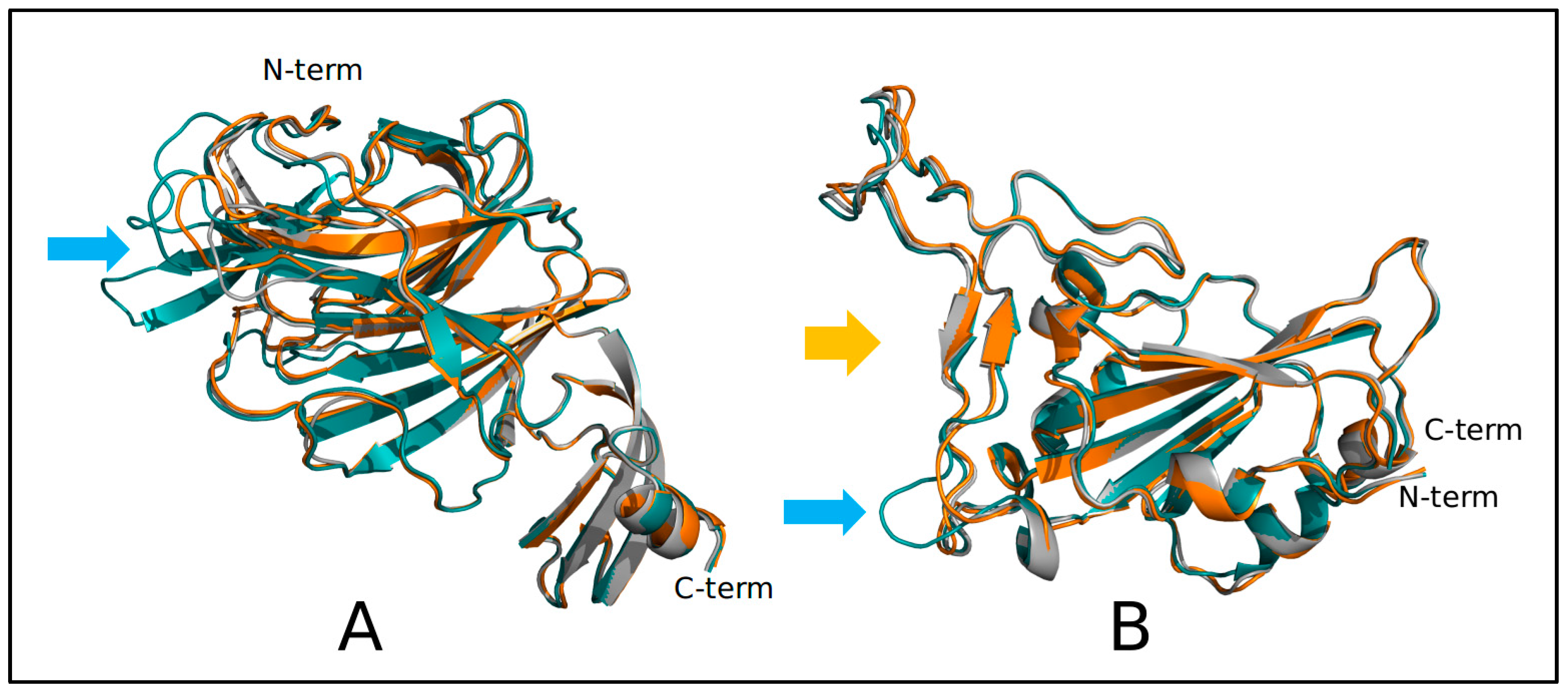
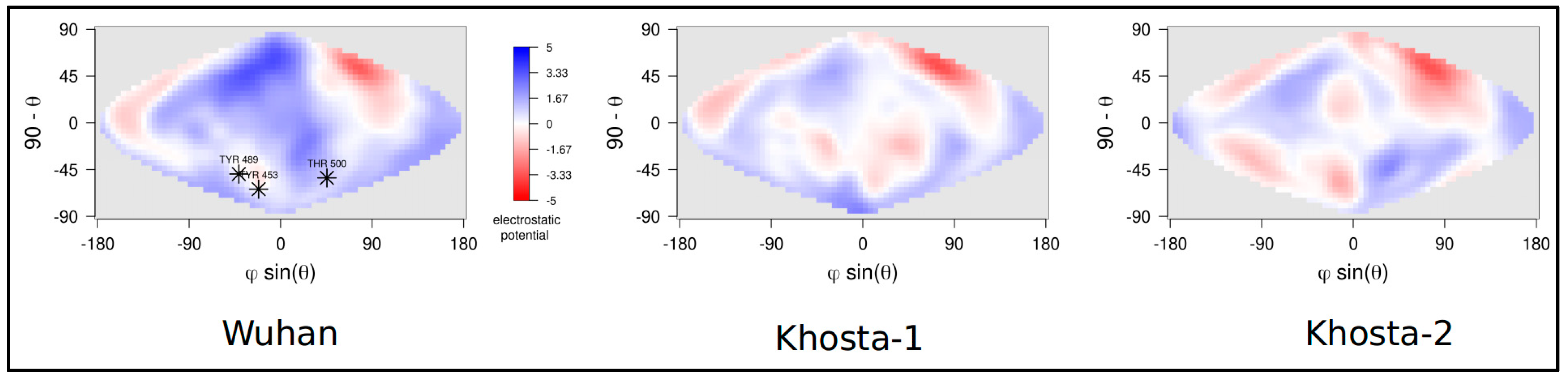
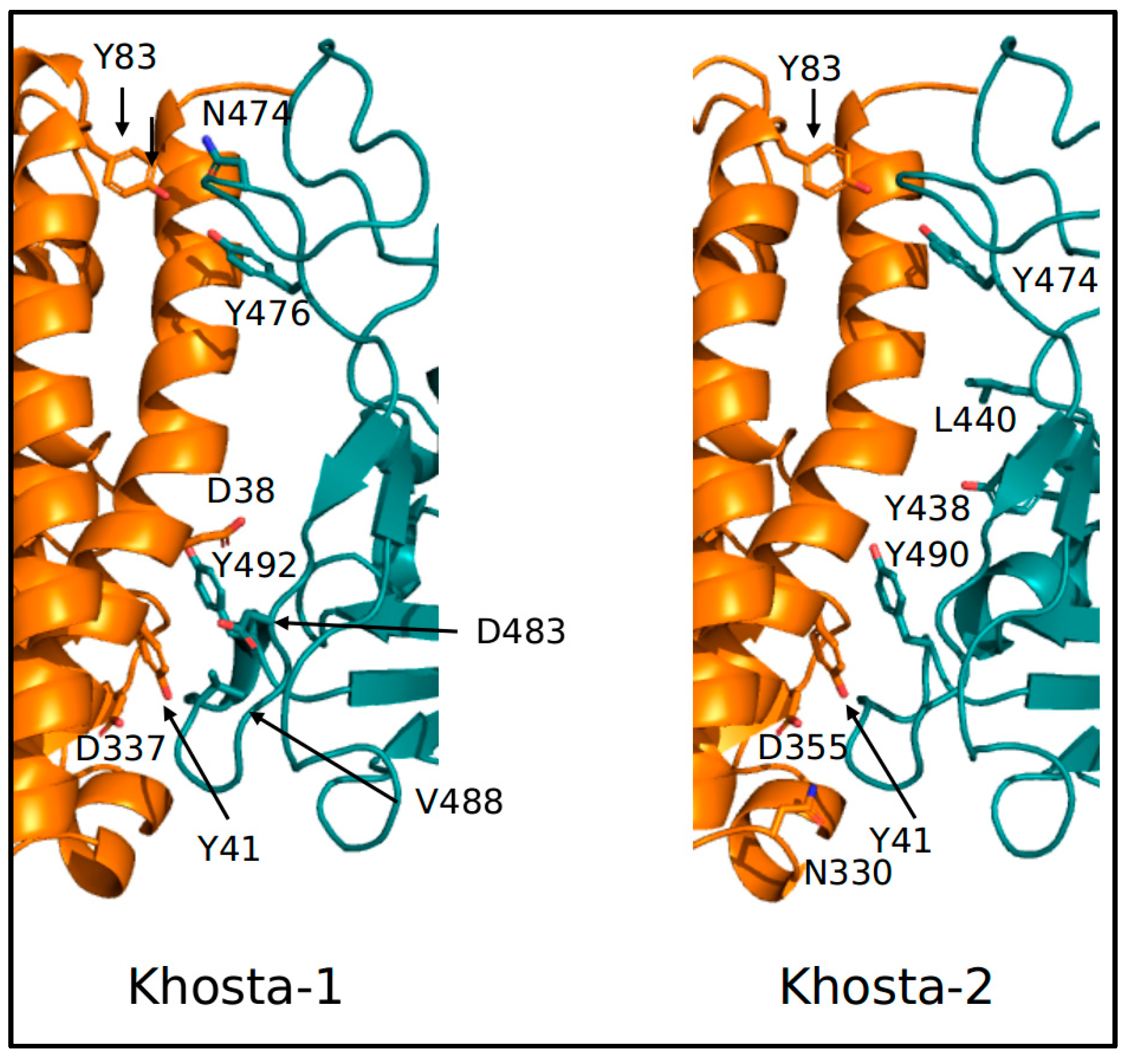
2.2. Structural and Molecular Dynamics Analyses
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roelle, S.M.; Shukla, N.; Pham, A.T.; Bruchez, A.M.; Matreyek, K.A. Expanded ACE2 dependencies of diverse SARS-like coronavirus receptor binding domains. PLoS Biol. 2022, 20, e3001738. [Google Scholar] [CrossRef] [PubMed]
- Alkhovsky, S.; Lenshin, S.; Romashin, A.; Vishnevskaya, T.; Vyshemirsky, O.; Bulycheva, Y.; Lvov, D.; Gitelman, A. SARS-like Coronaviruses in Horseshoe Bats (Rhinolophus spp.) in Russia, 2020. Viruses 2022, 14, 113. [Google Scholar] [CrossRef]
- Seifert, S.N.; Bai, S.; Fawcett, S.; Norton, E.B.; Zwezdaryk, K.J.; Robinson, J.; Gunn, B.; Letko, M. An ACE2-dependent Sarbecovirus in Russian bats is resistant to SARS-CoV-2 vaccines. PLoS Pathog. 2022, 18, e1010828. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Zhou, H.; Ji, J.; Chen, X.; Bi, Y.; Li, J.; Wang, Q.; Hu, T.; Song, H.; Zhao, R.; Chen, Y.; et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 2021, 184, 4380–4391.e14. [Google Scholar] [CrossRef]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chrétien, D.; Sanamxay, D.; et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 2022, 604, 330–336. [Google Scholar] [CrossRef]
- Yang, X.L.; Hu, B.; Wang, B.; Wang, M.N.; Zhang, Q.; Zhang, W.; Wu, L.J.; Ge, X.Y.; Zhang, Y.Z.; Daszak, P.; et al. Isolation and Characterization of a Novel Bat Coronavirus Closely Related to the Direct Progenitor of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2015, 90, 3253–3256. [Google Scholar] [CrossRef]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L., Jr.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Zepeda, S.K.; Walls, A.C.; Greaney, A.J.; Alkhovsky, S.; Veesler, D.; Bloom, J.D. ACE2 binding is an ancestral and evolvable trait of sarbecoviruses. Nature 2022, 603, 913–918. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Azzena, I.; Cossu, P.; Locci, C.; Angeletti, S.; Maruotti, A.; Ceccarelli, G.; Casu, M.; Fiori, P.L.; et al. Genetic Variability of the Monkeypox Virus Clade IIb B.1. J. Clin. Med. 2022, 11, 6388. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2017, 1653, 39–54. [Google Scholar]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System; Version 1.8; 2015. [Google Scholar]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2019, 7, 525–537. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. Publ. Protein Soc. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Schweke, H.; Mucchielli, M.-H.; Chevrollier, N.; Gosset, S.; Lopes, A. SURFMAP: A Software for Mapping in Two Dimensions Protein Surface Features. J. Chem. Inf. Model. 2022, 62, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Krüger, D.M.; Gohlke, H. DrugScorePPI webserver: Fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 2010, 38, W480–W486. [Google Scholar] [CrossRef]
- Cantoni, D.; Murray, M.J.; Kalemera, M.D.; Dicken, S.J.; Stejskal, L.; Brown, G.; Grove, J.; Lytras, S.; Coey, J.D.; McKenna, J.; et al. Evolutionary remodelling of N-terminal domain loops fine-tunes SARS-CoV-2 spike. EMBO Rep. 2022, 23, e54322. [Google Scholar] [CrossRef]
- Awasthi, M.; Gulati, S.; Sarkar, D.P.; Tiwari, S.; Kateriya, S.; Ranjan, P.; Verma, S.K. The sialoside-binding pocket of SARS-CoV-2 spike glycoprotein structurally resembles MERS-CoV. Viruses 2020, 12, 909. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Frost, S.D.W.; Magalis, B.R.; Pond, S.L.K. Neutral Theory and Rapidly Evolving Viral Pathogens. Mol. Biol. Evol. 2018, 35, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Ho, D.D.; Abdool Karim, Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet 2006, 368, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Sanna, D.; Azzena, I.; Cossu, P.; Giovanetti, M.; Benvenuto, D.; Coradduzza, E.; Alexiev, I.; Casu, M.; Fiori, P.L.; et al. Update on the Phylodynamics of SADS-CoV. Life 2021, 11, 820. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Giovanetti, M.; Pascarella, S.; Casu, M.; Ciccozzi, M. Avian influenza A H5N1: Are we really sure it is a spillover? Pathog. Glob. Health 2023, 117, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Mugosa, B.; Vujosevic, D.; Ciccozzi, M.; Valli, M.B.; Capobianchi, M.R.; Lo Presti, A.; Cella, E.; Giovanetti, M.; Lai, A.; Angeletti, S.; et al. Genetic diversity of the haemagglutinin (HA) of human influenza A (H1N1) virus in Montenegro: Focus on its origin and evolution. J. Med. Virol. 2016, 88, 1905–1913. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Sanna, D.; Azzena, I.; Casu, M.; Cossu, P.; Fiori, P.L.; Benvenuto, D.; Imperia, E.; Giovanetti, M.; Ceccarelli, G.; et al. Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages. J. Med. Virol. 2023, 95, e28625. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Khosta-1 | Khosta-2 | Wuhan | |
|---|---|---|---|
| Khosta-1 | - | 64 | 44 |
| Khosta-2 | 79 | - | 46 |
| Wuhan | 70 | 68 | - |
| Khosta-1 a | Khosta-2 a | Wuhan b | |
|---|---|---|---|
| RBD | 0.08 ± 0.01 | 0.79 ± 0.01 | 2.13 |
| NTD | 1.14 ± 0.05 | 4.89 ± 004 | 1.50 |
| Method | Khosta-1 | Khosta-2 | Wuhan |
|---|---|---|---|
| FoldX 5.0 | −12.00 | −9.40 | −16.88 |
| Mm/GBSA | −42.14 | −48.81 | −65.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpa, F.; Imperia, E.; Ciccozzi, A.; Pascarella, S.; Quaranta, M.; Giovanetti, M.; Borsetti, A.; Petrosillo, N.; Ciccozzi, M. Khosta: A Genetic and Structural Point of View of the Forgotten Virus. Infect. Dis. Rep. 2023, 15, 307-318. https://doi.org/10.3390/idr15030031
Scarpa F, Imperia E, Ciccozzi A, Pascarella S, Quaranta M, Giovanetti M, Borsetti A, Petrosillo N, Ciccozzi M. Khosta: A Genetic and Structural Point of View of the Forgotten Virus. Infectious Disease Reports. 2023; 15(3):307-318. https://doi.org/10.3390/idr15030031
Chicago/Turabian StyleScarpa, Fabio, Elena Imperia, Alessandra Ciccozzi, Stefano Pascarella, Miriana Quaranta, Marta Giovanetti, Alessandra Borsetti, Nicola Petrosillo, and Massimo Ciccozzi. 2023. "Khosta: A Genetic and Structural Point of View of the Forgotten Virus" Infectious Disease Reports 15, no. 3: 307-318. https://doi.org/10.3390/idr15030031
APA StyleScarpa, F., Imperia, E., Ciccozzi, A., Pascarella, S., Quaranta, M., Giovanetti, M., Borsetti, A., Petrosillo, N., & Ciccozzi, M. (2023). Khosta: A Genetic and Structural Point of View of the Forgotten Virus. Infectious Disease Reports, 15(3), 307-318. https://doi.org/10.3390/idr15030031