Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections



{kind=link}
Abstract
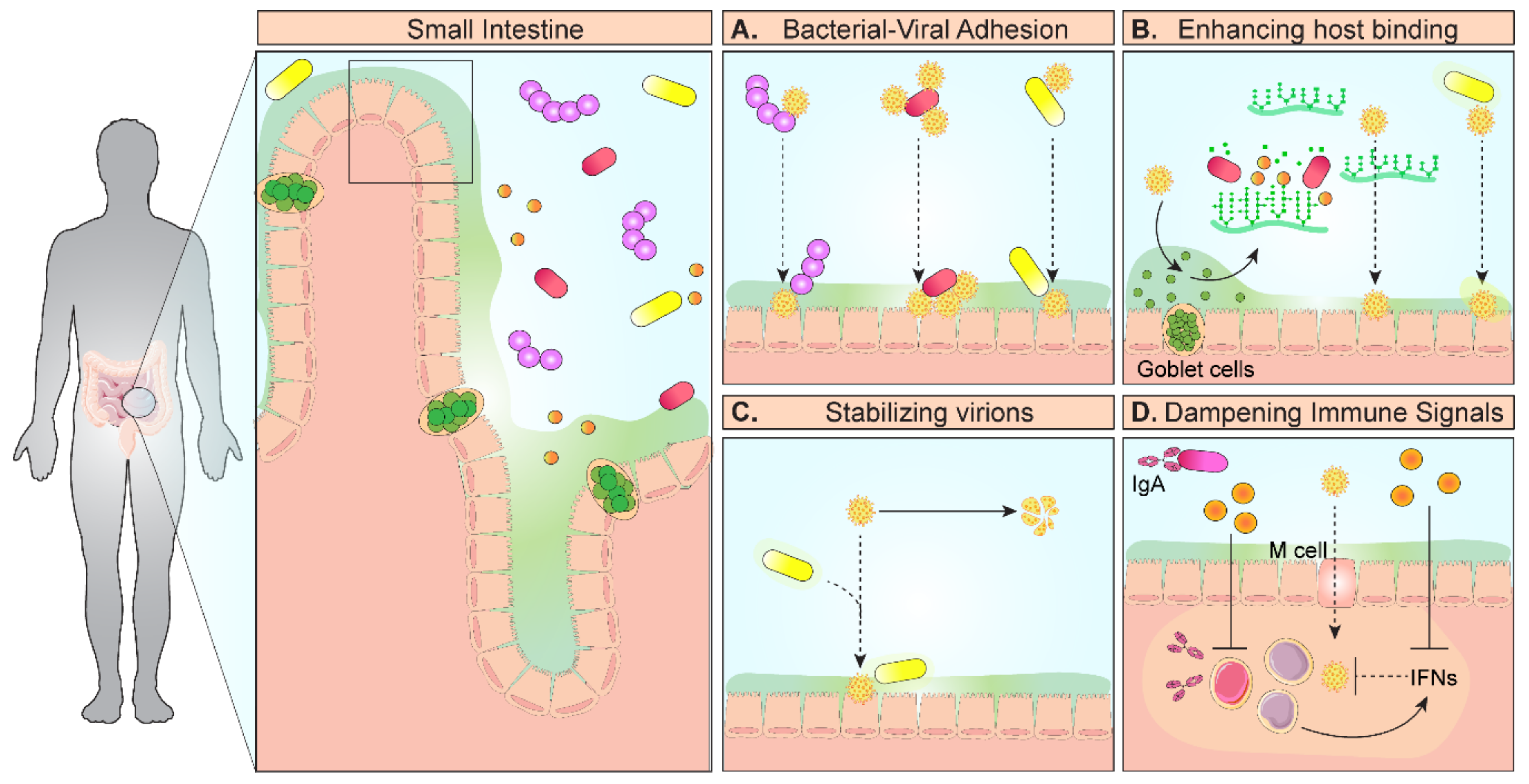
1. Introduction
2. Rotavirus (Family: Reoviridae, Genus: Rotavirus)
3. Norovirus (Family: Caliciviridae, Genus: Norovirus)
4. Poliovirus (Family: Picornaviridae, Genus: Poliovirus)
5. Reovirus (Family: Reoviridae, Genus: Orthoreovirus)
6. Astrovirus (Family: Astroviridae, Genus: Astrovirus)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Erickson, A.K.; Jesudhasan, P.R.; Mayer, M.J.; Narbad, A.; Winter, S.E.; Pfeiffer, J.K. Bacteria Facilitate Enteric Virus Co-infection of Mammalian Cells and Promote Genetic Recombination. Cell Host Microbe 2018, 23, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; Nice, T.J.; McCune, B.T.; Yokoyama, C.C.; Kambal, A.; Wheadon, M.; Diamond, M.S.; Ivanova, Y.; Artyomov, M.; Virgin, H.W. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 2015, 347, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Case, L.K.; Kopaskie, K.; Kozlova, A.; MacDearmid, C.; Chervonsky, A.V.; Golovkina, T.V. Successful transmission of a retrovirus depends on the commensal microbiota. Science 2011, 334, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Etheredge, C.A.; Pfeiffer, J.K. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog. 2008, 4, e1000082. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Jesudhasan, P.R.; Pfeiffer, J.K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 2014, 15, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, R.; Chassaing, B.; Zhang, B.; Gewirtz, A.T. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J. Infect. Dis. 2014, 210, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Banks, L.D.; Engevik, K.A.; Chang-Graham, A.L.; Perry, J.L.; Hutchinson, D.S.; Ajami, N.J.; Petrosino, J.F.; Hyser, J.M. Rotavirus infection induces glycan availability to promote ileum-specific changes in the microbiome aiding rotavirus virulence. Gut Microbes 2020, 11, 1324–1347. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Virgin, H.W. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 2016, 351. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Sonnenburg, J.L. The intestinal microbiota and viral susceptibility. Front. Microbiol. 2011, 2, 92. [Google Scholar] [CrossRef] [PubMed]
- Almand, E.A.; Moore, M.D.; Jaykus, L.A. Virus-Bacteria Interactions: An Emerging Topic in Human Infection. Viruses 2017, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea Among Children Younger Than 5 Years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef]
- Bomsel, M.; Alfsen, A. Entry of viruses through the epithelial barrier: Pathogenic trickery. Nat. Rev. Mol. Cell Biol. 2003, 4, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, S.; Woodruff, A.L.; Xia, M.; Tan, M.; Kennedy, M.A.; Jiang, X. Structural basis of glycan specificity of P[19] VP8*: Implications for rotavirus zoonosis and evolution. PLoS Pathog. 2017, 13, e1006707. [Google Scholar] [CrossRef]
- Pang, L.L.; Wang, M.X.; Sun, X.M.; Yuan, Y.; Qing, Y.; Xin, Y.; Zhang, J.Y.; Li, D.D.; Duan, Z.J. Glycan binding patterns of human rotavirus P[10] VP8* protein. Virol. J. 2018, 15, 161. [Google Scholar] [CrossRef]
- Jolly, C.L.; Beisner, B.M.; Holmes, I.H. Rotavirus infection of MA104 cells is inhibited by Ricinus lectin and separately expressed single binding domains. Virology 2000, 275, 89–97. [Google Scholar] [CrossRef][Green Version]
- Tailford, L.E.; Crost, E.H.; Kavanaugh, D.; Juge, N. Mucin glycan foraging in the human gut microbiome. Front. Genet. 2015, 6, 81. [Google Scholar] [CrossRef]
- Sun, X.; Li, D.; Qi, J.; Chai, W.; Wang, L.; Wang, L.; Peng, R.; Wang, H.; Zhang, Q.; Pang, L.; et al. Glycan Binding Specificity and Mechanism of Human and Porcine P[6]/P[19] Rotavirus VP8*s. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Boshuizen, J.A.; Reimerink, J.H.; Korteland-Van Male, A.M.; Van Ham, V.J.; Bouma, J.; Gerwig, G.J.; Koopmans, M.P.; Buller, H.A.; Dekker, J.; Einerhand, A.W. Homeostasis and function of goblet cells during rotavirus infection in mice. Virology 2005, 337, 210–221. [Google Scholar] [CrossRef]
- Chen, C.C.; Baylor, M.; Bass, D.M. Murine intestinal mucins inhibit rotavirus infection. Gastroenterology 1993, 105, 84–92. [Google Scholar] [CrossRef]
- Twitchell, E.L.; Tin, C.; Wen, K.; Zhang, H.; Becker-Dreps, S.; Azcarate-Peril, M.A.; Vilchez, S.; Li, G.; Ramesh, A.; Weiss, M.; et al. Modeling human enteric dysbiosis and rotavirus immunity in gnotobiotic pigs. Gut Pathog. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Vlasova, A.N.; Deblais, L.; Huang, H.C.; Wijeratne, A.; Kandasamy, S.; Fischer, D.D.; Langel, S.N.; Paim, F.C.; Alhamo, M.A.; et al. Impact of nutrition and rotavirus infection on the infant gut microbiota in a humanized pig model. BMC Gastroenterol. 2018, 18, 93. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Tsai, C.N.; Lee, Y.S.; Lin, C.Y.; Huang, K.Y.; Chao, H.C.; Lai, M.W.; Chiu, C.H. Intestinal microbiome in children with severe and complicated acute viral gastroenteritis. Sci. Rep. 2017, 7, 46130. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, M.; Zhang, C.; Du, H.; Wei, G.; Pang, X.; Zhou, H.; Liu, B.; Zhao, L. Pattern extraction of structural responses of gut microbiota to rotavirus infection via multivariate statistical analysis of clone library data. FEMS Microbiol. Ecol. 2009, 70, 21–29. [Google Scholar] [CrossRef]
- Shi, Z.; Zou, J.; Zhang, Z.; Zhao, X.; Noriega, J.; Zhang, B.; Zhao, C.; Ingle, H.; Bittinger, K.; Mattei, L.M.; et al. Segmented Filamentous Bacteria Prevent and Cure Rotavirus Infection. Cell 2019, 179, 644–685.e613. [Google Scholar] [CrossRef]
- Rodriguez-Diaz, J.; Garcia-Mantrana, I.; Vila-Vicent, S.; Gozalbo-Rovira, R.; Buesa, J.; Monedero, V.; Collado, M.C. Relevance of secretor status genotype and microbiota composition in susceptibility to rotavirus and norovirus infections in humans. Sci. Rep. 2017, 7, 45559. [Google Scholar] [CrossRef]
- Rahaman, M.M.; Wahed, M.A. Direct Nutrient Loss and Diarrhea. In Diarrhea and Malnutrition: Interactions, Mechanisms, and Interventions; Chen, L.C., Scrimshaw, N.S., Eds.; Springer: Boston, MA, USA, 1983; pp. 155–160. [Google Scholar] [CrossRef]
- Kandasamy, S.; Vlasova, A.N.; Fischer, D.; Kumar, A.; Chattha, K.S.; Rauf, A.; Shao, L.; Langel, S.N.; Rajashekara, G.; Saif, L.J. Differential Effects of Escherichia coli Nissle and Lactobacillus rhamnosus Strain GG on Human Rotavirus Binding, Infection, and B Cell Immunity. J. Immunol. 2016, 196, 1780–1789. [Google Scholar] [CrossRef]
- Greenberg, H.B.; Estes, M.K. Rotaviruses: From pathogenesis to vaccination. Gastroenterology 2009, 136, 1939–1951. [Google Scholar] [CrossRef]
- Blutt, S.E.; Miller, A.D.; Salmon, S.L.; Metzger, D.W.; Conner, M.E. IgA is important for clearance and critical for protection from rotavirus infection. Mucosal Immunol. 2012, 5, 712–719. [Google Scholar] [CrossRef]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.D.; Feng, N.; Sen, A.; Balan, M.; Tseng, H.C.; McElrath, C.; Smirnov, S.V.; Peng, J.; Yasukawa, L.L.; Durbin, R.K.; et al. Distinct Roles of Type I and Type III Interferons in Intestinal Immunity to Homologous and Heterologous Rotavirus Infections. PLoS Pathog. 2016, 12, e1005600. [Google Scholar] [CrossRef]
- Hernandez, P.P.; Mahlakoiv, T.; Yang, I.; Schwierzeck, V.; Nguyen, N.; Guendel, F.; Gronke, K.; Ryffel, B.; Hoelscher, C.; Dumoutier, L.; et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 2015, 16, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Rothenberg, M.E.; Mukherjee, G.; Feng, N.; Kalisky, T.; Nair, N.; Johnstone, I.M.; Clarke, M.F.; Greenberg, H.B. Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proc. Natl. Acad. Sci. USA 2012, 109, 20667–20672. [Google Scholar] [CrossRef]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef]
- Swiecki, M.; Miller, H.L.; Sesti-Costa, R.; Cella, M.; Gilfillan, S.; Colonna, M. Microbiota induces tonic CCL2 systemic levels that control pDC trafficking in steady state. Mucosal Immunol. 2017, 10, 936–945. [Google Scholar] [CrossRef]
- Vashist, S.; Bailey, D.; Putics, A.; Goodfellow, I. Model systems for the study of human norovirus Biology. Future Virol. 2009, 4, 353–367. [Google Scholar] [CrossRef]
- Payne, D.C.; Vinje, J.; Szilagyi, P.G.; Edwards, K.M.; Staat, M.A.; Weinberg, G.A.; Hall, C.B.; Chappell, J.; Bernstein, D.I.; Curns, A.T.; et al. Norovirus and medically attended gastroenteritis in U.S. children. N. Engl. J. Med. 2013, 368, 1121–1130. [Google Scholar] [CrossRef]
- Karst, S.M.; Wobus, C.E. A working model of how noroviruses infect the intestine. PLoS Pathog. 2015, 11, e1004626. [Google Scholar] [CrossRef]
- Patel, M.M.; Widdowson, M.A.; Glass, R.I.; Akazawa, K.; Vinje, J.; Parashar, U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008, 14, 1224–1231. [Google Scholar] [CrossRef]
- Estes, M.K.; Ettayebi, K.; Tenge, V.R.; Murakami, K.; Karandikar, U.; Lin, S.C.; Ayyar, B.V.; Cortes-Penfield, N.W.; Haga, K.; Neill, F.H.; et al. Human Norovirus Cultivation in Nontransformed Stem Cell-Derived Human Intestinal Enteroid Cultures: Success and Challenges. Viruses 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Grau, K.R.; Zhu, S.; Peterson, S.T.; Helm, E.W.; Philip, D.; Phillips, M.; Hernandez, A.; Turula, H.; Frasse, P.; Graziano, V.R.; et al. The intestinal regionalization of acute norovirus infection is regulated by the microbiota via bile acid-mediated priming of type III interferon. Nat. Microbiol. 2020, 5, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Twitchell, E.L.; Ramesh, A.K.; Bui, T.; Majette, E.; Tin, C.M.; Avery, R.; Arango-Argoty, G.; Zhang, L.; Becker-Dreps, S.; et al. Enhanced GII.4 human norovirus infection in gnotobiotic pigs transplanted with a human gut microbiota. J. Gen. Virol. 2019, 100, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Brandl, M.; Mandrell, R. Porcine gastric mucin binds to recombinant norovirus particles and competitively inhibits their binding to histo-blood group antigens and Caco-2 cells. Lett. Appl. Microbiol. 2005, 41, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Yang, D.; Jiang, X.; Zhong, W.; Cannon, J.L.; Burkhardt, W., 3rd; Woods, J.W.; Hartman, G.; Lindesmith, L.; Baric, R.S.; et al. Specificity and kinetics of norovirus binding to magnetic bead-conjugated histo-blood group antigens. J. Appl. Microbiol. 2010, 109, 1753–1762. [Google Scholar] [CrossRef]
- Li, X.; Chen, H. Evaluation of the porcine gastric mucin binding assay for high-pressure-inactivation studies using murine norovirus and tulane virus. Appl. Environ. Microbiol. 2015, 81, 515–521. [Google Scholar] [CrossRef]
- Nelson, A.M.; Walk, S.T.; Taube, S.; Taniuchi, M.; Houpt, E.R.; Wobus, C.E.; Young, V.B. Disruption of the human gut microbiota following Norovirus infection. PLoS ONE 2012, 7, e48224. [Google Scholar] [CrossRef]
- Walker, F.C.; Baldridge, M.T. Interactions between noroviruses, the host, and the microbiota. Curr. Opin. Virol. 2019, 37, 1–9. [Google Scholar] [CrossRef]
- Patin, N.V.; Pena-Gonzalez, A.; Hatt, J.K.; Moe, C.; Kirby, A.; Konstantinidis, K.T. The Role of the Gut Microbiome in Resisting Norovirus Infection as Revealed by a Human Challenge Study. mBio 2020, 11. [Google Scholar] [CrossRef]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Turula, H.; Bragazzi Cunha, J.; Mainou, B.A.; Ramakrishnan, S.K.; Wilke, C.A.; Gonzalez-Hernandez, M.B.; Pry, A.; Fava, J.; Bassis, C.M.; Edelman, J.; et al. Natural Secretory Immunoglobulins Promote Enteric Viral Infections. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Breiman, A.; Le Pendu, J.; Uyttendaele, M. Binding to histo-blood group antigen-expressing bacteria protects human norovirus from acute heat stress. Front. Microbiol. 2015, 6, 659. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Sano, D.; Suenaga, A.; Yoshimura, T.; Fuzawa, M.; Nakagomi, T.; Nakagomi, O.; Okabe, S. Histo-blood group antigen-like substances of human enteric bacteria as specific adsorbents for human noroviruses. J. Virol. 2013, 87, 9441–9451. [Google Scholar] [CrossRef] [PubMed]
- Almand, E.A.; Moore, M.D.; Outlaw, J.; Jaykus, L.A. Human norovirus binding to select bacteria representative of the human gut microbiota. PLoS ONE 2017, 12, e0173124. [Google Scholar] [CrossRef]
- Karst, S.M. The influence of commensal bacteria on infection with enteric viruses. Nat. Rev. Microbiol. 2016, 14, 197–204. [Google Scholar] [CrossRef]
- Kilic, T.; Koromyslova, A.; Hansman, G.S. Structural Basis for Human Norovirus Capsid Binding to Bile Acids. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Nelson, C.A.; Wilen, C.B.; Dai, Y.N.; Orchard, R.C.; Kim, A.S.; Stegeman, R.A.; Hsieh, L.L.; Smith, T.J.; Virgin, H.W.; Fremont, D.H. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc. Natl. Acad. Sci. USA 2018, 115, E9201–E9210. [Google Scholar] [CrossRef]
- Murakami, K.; Tenge, V.R.; Karandikar, U.C.; Lin, S.C.; Ramani, S.; Ettayebi, K.; Crawford, S.E.; Zeng, X.L.; Neill, F.H.; Ayyar, B.V.; et al. Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication in human intestinal enteroids. Proc. Natl. Acad. Sci. USA 2020, 117, 1700–1710. [Google Scholar] [CrossRef]
- Mallory, M.L.; Lindesmith, L.C.; Brewer-Jensen, P.D.; Graham, R.L.; Baric, R.S. Bile Facilitates Human Norovirus Interactions with Diverse Histoblood Group Antigens, Compensating for Capsid Microvariation Observed in 2016–2017 GII.2 Strains. Viruses 2020, 12, 989. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, T.Y.; Kim, Y.; Lee, S.H.; Kim, S.; Kang, S.W.; Yang, J.Y.; Baek, I.J.; Sung, Y.H.; Park, Y.Y.; et al. Microbiota-Derived Lactate Accelerates Intestinal Stem-Cell-Mediated Epithelial Development. Cell Host Microbe 2018, 24, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Luk, B.; Chang-Graham, A.L.; Hall, A.; Herrmann, B.; Ruan, W.; Endres, B.T.; Shi, Z.; Garey, K.W.; Hyser, J.M.; et al. Bifidobacterium dentium Fortifies the Intestinal Mucus Layer via Autophagy and Calcium Signaling Pathways. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Troll, J.V.; Hamilton, M.K.; Abel, M.L.; Ganz, J.; Bates, J.M.; Stephens, W.Z.; Melancon, E.; Van der Vaart, M.; Meijer, A.H.; Distel, M.; et al. Microbiota promote secretory cell determination in the intestinal epithelium by modulating host Notch signaling. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.C.E.; Shanahan, M.T.; Singh, A.P.; Sethupathy, P. Gut Microbial Influences on the Mammalian Intestinal Stem Cell Niche. Stem Cells Int. 2017, 2017, 5604727. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Lee, S.; Hsieh, L.L.; Orchard, R.C.; Desai, C.; Hykes, B.L., Jr.; McAllaster, M.R.; Balce, D.R.; Feehley, T.; Brestoff, J.R.; et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 2018, 360, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Izawa, K.; Kitaura, J.; Yamanishi, Y.; Matsuoka, T.; Oki, T.; Shibata, F.; Kumagai, H.; Nakajima, H.; Maeda-Yamamoto, M.; Hauchins, J.P.; et al. Functional analysis of activating receptor LMIR4 as a counterpart of inhibitory receptor LMIR3. J. Biol. Chem. 2007, 282, 17997–18008. [Google Scholar] [CrossRef]
- Hogle, J.M. Poliovirus cell entry: Common structural themes in viral cell entry pathways. Annu. Rev. Microbiol. 2002, 56, 677–702. [Google Scholar] [CrossRef]
- Zarocostas, J. WHO keeps polio on the international health emergency list. Lancet 2018, 392, 2425. [Google Scholar] [CrossRef]
- Stower, H. WHO declares Africa polio-free. Nat. Med. 2020, 26, 1805. [Google Scholar] [CrossRef]
- Berger, A.K.; Yi, H.; Kearns, D.B.; Mainou, B.A. Bacteria and bacterial envelope components enhance mammalian reovirus thermostability. PLoS Pathog. 2017, 13, e1006768. [Google Scholar] [CrossRef]
- Ouattara, L.A.; Barin, F.; Barthez, M.A.; Bonnaud, B.; Roingeard, P.; Goudeau, A.; Castelnau, P.; Vernet, G.; Paranhos-Baccala, G.; Komurian-Pradel, F. Novel human reovirus isolated from children with acute necrotizing encephalopathy. Emerg. Infect. Dis. 2011, 17, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Tai, J.H.; Williams, J.V.; Edwards, K.M.; Wright, P.F.; Crowe, J.E., Jr.; Dermody, T.S. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J. Infect. Dis. 2005, 191, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L. Serologic grouping of reoviruses by hemagglutination-inhibition. Am. J. Hyg. 1960, 71, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Antar, A.A.; Konopka, J.L.; Campbell, J.A.; Henry, R.A.; Perdigoto, A.L.; Carter, B.D.; Pozzi, A.; Abel, T.W.; Dermody, T.S. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 2009, 5, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.H.; Eaton, M.A.; Anderson, A.O. Reovirus infection in adult mice: The virus hemagglutinin determines the site of intestinal disease. Microb. Pathog. 1986, 1, 79–87. [Google Scholar] [CrossRef]
- Amerongen, H.M.; Wilson, G.A.; Fields, B.N.; Neutra, M.R. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J. Virol. 1994, 68, 8428–8432. [Google Scholar] [CrossRef]
- Vu, D.L.; Bosch, A.; Pinto, R.M.; Guix, S. Epidemiology of Classic and Novel Human Astrovirus: Gastroenteritis and Beyond. Viruses 2017, 9, 33. [Google Scholar] [CrossRef]
- Olortegui, M.P.; Rouhani, S.; Yori, P.P.; Salas, M.S.; Trigoso, D.R.; Mondal, D.; Bodhidatta, L.; Platts-Mills, J.; Samie, A.; Kabir, F.; et al. Astrovirus Infection and Diarrhea in 8 Countries. Pediatrics 2018, 141. [Google Scholar] [CrossRef]
- GBD Diarrhoeal Diseases Collaborators. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis. 2017, 17, 909–948. [Google Scholar] [CrossRef]
- Cortez, V.; Meliopoulos, V.A.; Karlsson, E.A.; Hargest, V.; Johnson, C.; Schultz-Cherry, S. Astrovirus Biology and Pathogenesis. Annu. Rev. Virol. 2017, 4, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rodriguez, F.J.; Vieille, G.; Turin, L.; Yildiz, S.; Tapparel, C.; Kaiser, L. Fecal Components Modulate Human Astrovirus Infectivity in Cells and Reconstituted Intestinal Tissues. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.; Boyd, D.F.; Crawford, J.C.; Sharp, B.; Livingston, B.; Rowe, H.M.; Davis, A.; Alsallaq, R.; Robinson, C.G.; Vogel, P.; et al. Astrovirus infects actively secreting goblet cells and alters the gut mucus barrier. Nat. Commun. 2020, 11, 2097. [Google Scholar] [CrossRef] [PubMed]
- Kolawole, A.O.; Mirabelli, C.; Hill, D.R.; Svoboda, S.A.; Janowski, A.B.; Passalacqua, K.D.; Rodriguez, B.N.; Dame, M.K.; Freiden, P.; Berger, R.P.; et al. Astrovirus replication in human intestinal enteroids reveals multi-cellular tropism and an intricate host innate immune landscape. PLoS Pathog. 2019, 15, e1008057. [Google Scholar] [CrossRef]
- Cortez, V.; Margolis, E.; Schultz-Cherry, S. Astrovirus and the microbiome. Curr. Opin. Virol. 2019, 37, 10–15. [Google Scholar] [CrossRef]
- Cortez, V.; Sharp, B.; Yao, J.; Livingston, B.; Vogel, P.; Schultz-Cherry, S. Characterizing a Murine Model for Astrovirus Using Viral Isolates from Persistently Infected Immunocompromised Mice. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Ma, C.; Wu, X.; Nawaz, M.; Li, J.; Yu, P.; Moore, J.E.; Xu, J. Molecular characterization of fecal microbiota in patients with viral diarrhea. Curr. Microbiol. 2011, 63, 259–266. [Google Scholar] [CrossRef]
- Qureshi, M.A.; Saif, Y.M.; Heggen-Peay, C.L.; Edens, F.W.; Havenstein, G.B. Induction of functional defects in macrophages by a poult enteritis and mortality syndrome-associated turkey astrovirus. Avian Dis. 2001, 45, 853–861. [Google Scholar] [CrossRef]
- Edens, F.W.; Parkhurst, C.R.; Qureshi, M.A.; Casas, I.A.; Havenstein, G.B. Atypical Escherichia coli strains and their association with poult enteritis and mortality syndrome. Poult. Sci. 1997, 76, 952–960. [Google Scholar] [CrossRef]
- Wasimuddin; Brandel, S.D.; Tschapka, M.; Page, R.; Rasche, A.; Corman, V.M.; Drosten, C.; Sommer, S. Astrovirus infections induce age-dependent dysbiosis in gut microbiomes of bats. ISME J. 2018, 12, 2883–2893. [Google Scholar] [CrossRef]
- Ingle, H.; Lee, S.; Ai, T.; Orvedahl, A.; Rodgers, R.; Zhao, G.; Sullender, M.; Peterson, S.T.; Locke, M.; Liu, T.C.; et al. Viral complementation of immunodeficiency confers protection against enteric pathogens via interferon-lambda. Nat. Microbiol. 2019, 4, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Willing, B.; Keeney, K.M.; Menendez, A.; Bergstrom, K.S.; Gill, N.; Russell, S.L.; Vallance, B.A.; Finlay, B.B. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect. Immun. 2011, 79, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, S.; Chattha, K.S.; Vlasova, A.N.; Rajashekara, G.; Saif, L.J. Lactobacilli and Bifidobacteria enhance mucosal B cell responses and differentially modulate systemic antibody responses to an oral human rotavirus vaccine in a neonatal gnotobiotic pig disease model. Gut Microbes 2014, 5, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.; Vimont, A.; Darveau, A.; Fliss, I.; Jean, J. Study of the Ability of Bifidobacteria of Human Origin to Prevent and Treat Rotavirus Infection Using Colonic Cell and Mouse Models. PLoS ONE 2016, 11, e0164512. [Google Scholar] [CrossRef]
- Ishizuka, T.; Kanmani, P.; Kobayashi, H.; Miyazaki, A.; Soma, J.; Suda, Y.; Aso, H.; Nochi, T.; Iwabuchi, N.; Xiao, J.Z.; et al. Immunobiotic Bifidobacteria Strains Modulate Rotavirus Immune Response in Porcine Intestinal Epitheliocytes via Pattern Recognition Receptor Signaling. PLoS ONE 2016, 11, e0152416. [Google Scholar] [CrossRef]
- Phuapradit, P.; Varavithya, W.; Vathanophas, K.; Sangchai, R.; Podhipak, A.; Suthutvoravut, U.; Nopchinda, S.; Chantraruksa, V.; Haschke, F. Reduction of rotavirus infection in children receiving bifidobacteria-supplemented formula. J. Med. Assoc. Thail. 1999, 82, S43–S48. [Google Scholar]
- Azagra-Boronat, I.; Massot-Cladera, M.; Knipping, K.; Garssen, J.; Ben Amor, K.; Knol, J.; Franch, A.; Castell, M.; Rodriguez-Lagunas, M.J.; Perez-Cano, F.J. Strain-Specific Probiotic Properties of Bifidobacteria and Lactobacilli for the Prevention of Diarrhea Caused by Rotavirus in a Preclinical Model. Nutrients 2020, 12, 498. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Kandasamy, S.; Chattha, K.S.; Rajashekara, G.; Saif, L.J. Comparison of probiotic lactobacilli and bifidobacteria effects, immune responses and rotavirus vaccines and infection in different host species. Vet. Immunol. Immunopathol. 2016, 172, 72–84. [Google Scholar] [CrossRef]
- Qiao, H.; Duffy, L.C.; Griffiths, E.; Dryja, D.; Leavens, A.; Rossman, J.; Rich, G.; Riepenhoff-Talty, M.; Locniskar, M. Immune responses in rhesus rotavirus-challenged BALB/c mice treated with bifidobacteria and prebiotic supplements. Pediatr. Res. 2002, 51, 750–755. [Google Scholar] [CrossRef]
- Lee, W.; Kim, M.; Lee, S.H.; Jung, H.G.; Oh, J.W. Prophylactic efficacy of orally administered Bacillus poly-gamma-glutamic acid, a non-LPS TLR4 ligand, against norovirus infection in mice. Sci. Rep. 2018, 8, 8667. [Google Scholar] [CrossRef]
- Lee, H.; Ko, G. Antiviral effect of vitamin A on norovirus infection via modulation of the gut microbiome. Sci. Rep. 2016, 6, 25835. [Google Scholar] [CrossRef] [PubMed]
- Long, K.Z.; Garcia, C.; Santos, J.I.; Rosado, J.L.; Hertzmark, E.; Dupont, H.L.; Ko, G. Vitamin A supplementation has divergent effects on norovirus infections and clinical symptoms among Mexican children. J. Infect. Dis. 2007, 196, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Roubaud-Baudron, C.; Ruiz, V.E.; Swan, A.M., Jr.; Vallance, B.A.; Ozkul, C.; Pei, Z.; Li, J.; Battaglia, T.W.; Perez-Perez, G.I.; Blaser, M.J. Long-Term Effects of Early-Life Antibiotic Exposure on Resistance to Subsequent Bacterial Infection. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, A.J.; Wang, B.; Sun, X.; Kennedy, E.A.; Hernandez-Leyva, A.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Dantas, G. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 2019, 4, 2285–2297. [Google Scholar] [CrossRef]
- Sbihi, H.; Boutin, R.C.; Cutler, C.; Suen, M.; Finlay, B.B.; Turvey, S.E. Thinking bigger: How early-life environmental exposures shape the gut microbiome and influence the development of asthma and allergic disease. Allergy 2019, 74, 2103–2115. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef]
- Lawson, M.A.E.; O’Neill, I.J.; Kujawska, M.; Gowrinadh Javvadi, S.; Wijeyesekera, A.; Flegg, Z.; Chalklen, L.; Hall, L.J. Breast milk-derived human milk oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J. 2020, 14, 635–648. [Google Scholar] [CrossRef]
- Thongaram, T.; Hoeflinger, J.L.; Chow, J.; Miller, M.J. Human milk oligosaccharide consumption by probiotic and human-associated bifidobacteria and lactobacilli. J. Dairy Sci. 2017, 100, 7825–7833. [Google Scholar] [CrossRef]
- Gotoh, A.; Katoh, T.; Sakanaka, M.; Ling, Y.; Yamada, C.; Asakuma, S.; Urashima, T.; Tomabechi, Y.; Katayama-Ikegami, A.; Kurihara, S.; et al. Sharing of human milk oligosaccharides degradants within bifidobacterial communities in faecal cultures supplemented with Bifidobacterium bifidum. Sci. Rep. 2018, 8, 13958. [Google Scholar] [CrossRef]
- Krammer, E.M.; Bouckaert, J.M.J. Norovirus devours human milk oligosaccharides rich in alpha-fucose. J. Biol. Chem. 2018, 293, 11966–11967. [Google Scholar] [CrossRef]
- Koromyslova, A.; Tripathi, S.; Morozov, V.; Schroten, H.; Hansman, G.S. Human norovirus inhibition by a human milk oligosaccharide. Virology 2017, 508, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Laucirica, D.R.; Triantis, V.; Schoemaker, R.; Estes, M.K.; Ramani, S. Milk Oligosaccharides Inhibit Human Rotavirus Infectivity in MA104 Cells. J. Nutr. 2017, 147, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Ramani, S.; Stewart, C.J.; Laucirica, D.R.; Ajami, N.J.; Robertson, B.; Autran, C.A.; Shinge, D.; Rani, S.; Anandan, S.; Hu, L.; et al. Human milk oligosaccharides, milk microbiome and infant gut microbiome modulate neonatal rotavirus infection. Nat. Commun. 2018, 9, 5010. [Google Scholar] [CrossRef] [PubMed]
- Chichlowski, M.; De Lartigue, G.; German, J.B.; Raybould, H.E.; Mills, D.A. Bifidobacteria isolated from infants and cultured on human milk oligosaccharides affect intestinal epithelial function. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.Y.; Li, B.; Koike, Y.; Maattanen, P.; Miyake, H.; Cadete, M.; Johnson-Henry, K.C.; Botts, S.R.; Lee, C.; Abrahamsson, T.R.; et al. Human Milk Oligosaccharides Increase Mucin Expression in Experimental Necrotizing Enterocolitis. Mol. Nutr. Food Res. 2019, 63, e1800658. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Kong, C.; Walvoort, M.T.C.; Faas, M.M.; De Vos, P. Human Milk Oligosaccharides Differently Modulate Goblet Cells Under Homeostatic, Proinflammatory Conditions and ER Stress. Mol. Nutr. Food Res. 2020, 64, e1900976. [Google Scholar] [CrossRef]
- Natividad, J.M.; Rytz, A.; Keddani, S.; Bergonzelli, G.; Garcia-Rodenas, C.L. Blends of Human Milk Oligosaccharides Confer Intestinal Epithelial Barrier Protection in Vitro. Nutrients 2020, 12, 3047. [Google Scholar] [CrossRef]
- Engevik, M.A.; Aihara, E.; Montrose, M.H.; Shull, G.E.; Hassett, D.J.; Worrell, R.T. Loss of NHE3 alters gut microbiota composition and influences Bacteroides thetaiotaomicron growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G697–G711. [Google Scholar] [CrossRef]
- Engevik, M.A.; Hickerson, A.; Shull, G.E.; Worrell, R.T. Acidic conditions in the NHE2(−/−) mouse intestine result in an altered mucosa-associated bacterial population with changes in mucus oligosaccharides. Cell Physiol. Biochem. 2013, 32, 111–128. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engevik, K.A.; Engevik, M.A. Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections. Gastroenterol. Insights 2021, 12, 41-55. https://doi.org/10.3390/gastroent12010005
Engevik KA, Engevik MA. Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections. Gastroenterology Insights. 2021; 12(1):41-55. https://doi.org/10.3390/gastroent12010005
Chicago/Turabian StyleEngevik, Kristen A., and Melinda A. Engevik. 2021. "Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections" Gastroenterology Insights 12, no. 1: 41-55. https://doi.org/10.3390/gastroent12010005
APA StyleEngevik, K. A., & Engevik, M. A. (2021). Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections. Gastroenterology Insights, 12(1), 41-55. https://doi.org/10.3390/gastroent12010005