
Brugada Syndrome: Channelopathy and/or Cardiomyopathy

 ,
, 
Abstract
1. Introduction
2. Electrocardiography and Invasive Electrophysiology: A Complex Interaction of Repolarization and Depolarization Abnormalities
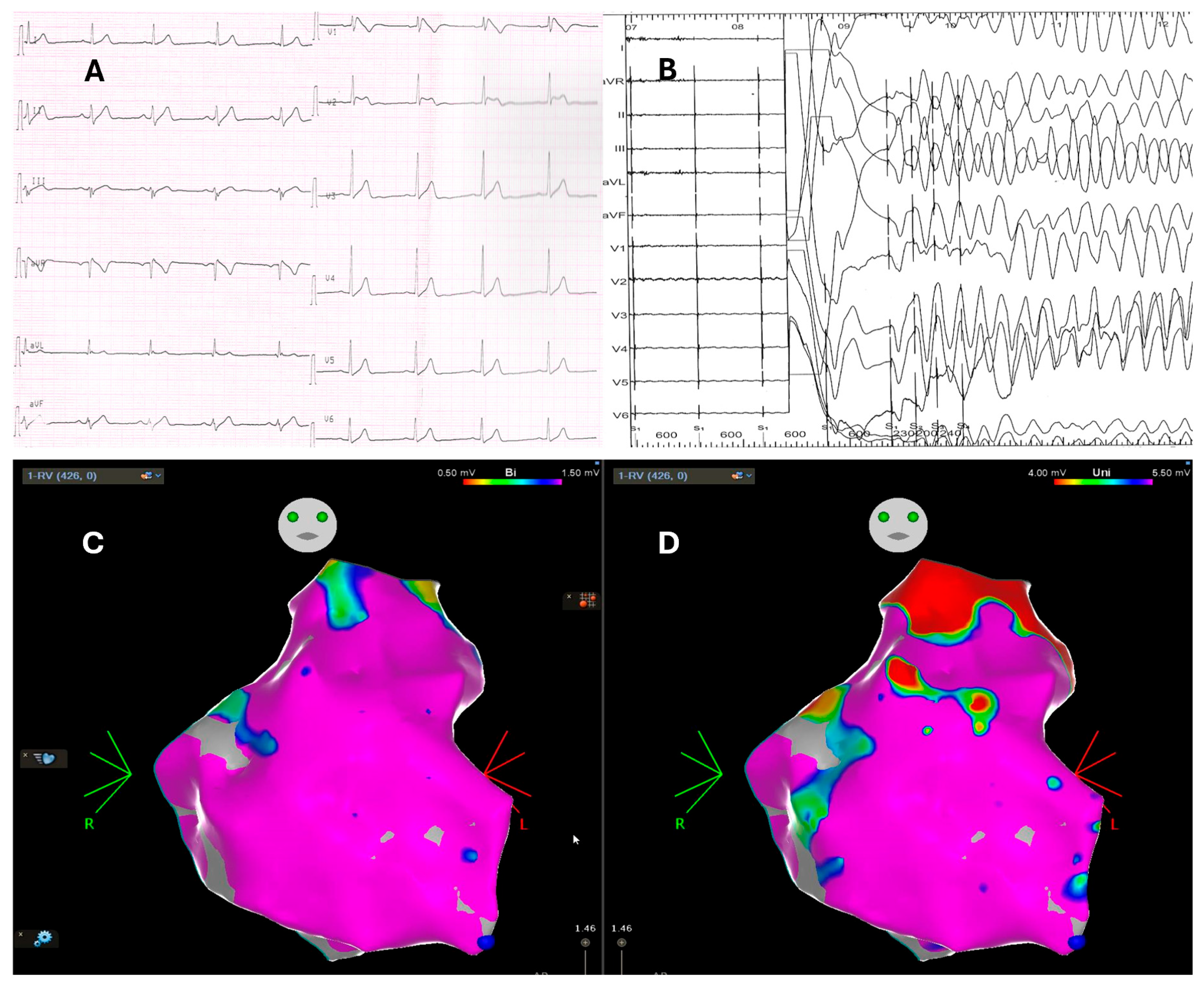
3. Electrophysiological Study: The Prognostic Role of Programmed Ventricular Stimulation
4. Multimodality Imaging: Subtle Functional Abnormalities Revealed by Advanced Echocardiography and Cardiac Magnetic Resonance
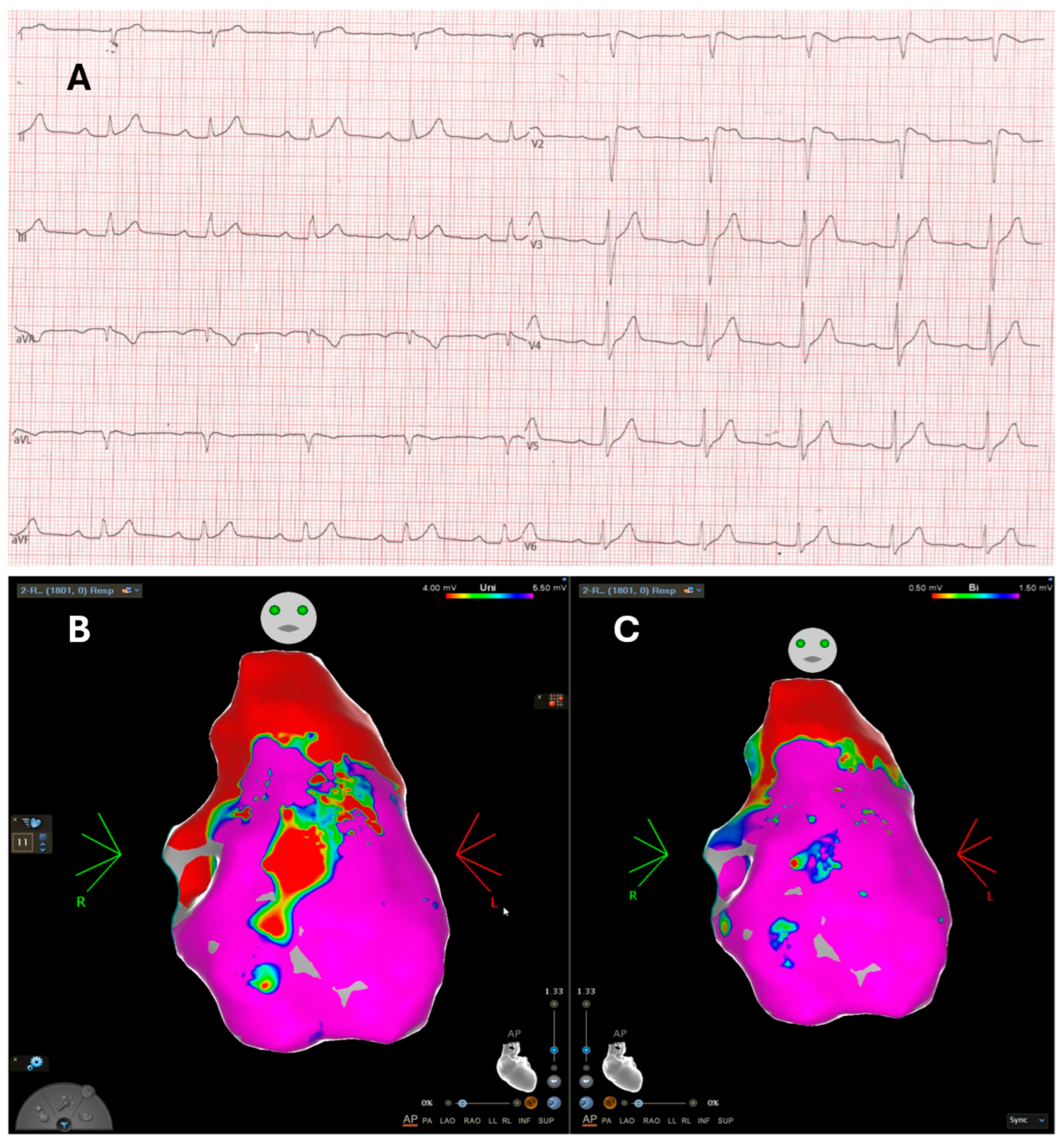
5. Electroanatomic Mapping: Electrical and Structural Substrate Insights
6. Histology and Immunology: Exploring the Substrate and the Role of the Immune System in the Disease
7. Brugada Syndrome and Arrhythmogenic Cardiomyopathy: A Common Spectrum of Disease
8. Future Perspectives: Genetics and Immune Modulation
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BrS | Brugada syndrome |
| EPS | Electrophysiological study |
| EAM | Electroanatomic mapping |
| LP | Late potential |
| SA-ECG | Signal-average ECG |
| f-QRS | Fragmented QRS |
| RV | Right ventricle |
| LV | Left ventricle |
| RVOT | Right ventricular outflow tract |
| AV | Atrioventricular block |
| VT | Ventricular tachycardia |
| VF | Ventricular fibrillation |
| PVS | Programmed ventricular stimulation |
| RIMP | Right ventricular index of myocardial performance |
| RVEF | Right ventricular ejection fraction |
| CMR | Cardiac magnetic resonance |
| LGE | Late gadolinium enhancement |
| RVOT-FT | Right ventricular outflow tract-feature tracking |
| EMB | Endomyocardial biopsy |
References
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Veerakul, G.; Nimmannit, S.; Chaowakul, V.; Bhuripanyo, K.; Likittanasombat, K.; Tunsanga, K.; Kuasirikul, S.; Malasit, P.; Tansupasawadikul, S.; et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 1997, 96, 2595–2600. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]
- Wilde, A.A.; Postema, P.G.; Di Diego, J.M.; Viskin, S.; Morita, H.; Fish, J.M.; Antzelevitch, C. The pathophysiological mechanism underlying Brugada syndrome. Depolarization versus repolarization. J. Mol. Cell. Cardiol. 2010, 49, 543–553. [Google Scholar] [CrossRef]
- Sieira, J.; Dendramis, G.; Brugada, P. Pathogenesis and management of Brugada syndrome. Nat. Rev. Cardiol. 2016, 13, 744–756. [Google Scholar] [CrossRef]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef]
- Sarquella-Brugada, G.; Campuzano, O.; Arbelo, E.; Brugada, J.; Brugada, R. Brugada syndrome: Clinical and genetic findings. Genet. Med. 2016, 18, 3–12. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef]
- Peters, S.; Thompson, B.A.; Perrin, M.; James, P.; Zentner, D.; Kalman, J.M.; Vandenberg, J.I.; Fatkin, D. Arrhythmic Phenotypes Are a Defining Feature of Dilated Cardiomyopathy-Associated SCN5A Variants: A Systematic Review. Circ. Genomic Precis. Med. 2022, 15, E003432. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Veerakul, G.; Chandanamattha, P.; Chaothawee, L.; Ariyachaipanich, A.; Jirasirirojanakorn, K.; Likittanasombat, K.; Bhuripanyo, K.; Ngarmukos, T. Prevention of ventricular fibrillation episodes in brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011, 123, 1270–1279. [Google Scholar] [CrossRef]
- Letsas, K.P.; Efremidis, M.; Asvestas, D.; Vlachos, K.; Georgopoulos, S.; Tse, G.; Liu, T.; Bazoukis, G.; Sideris, A.; Baranchuk, A.; et al. Right Ventricular Outflow Tract Electroanatomical Abnormalities Predict Ventricular Fibrillation Inducibility in Brugada Syndrome. Circ. Arrhythm. Electrophysiol. 2018, 11, e005928. [Google Scholar] [CrossRef]
- Frustaci, A.; Priori, S.G.; Pieroni, M.; Chimenti, C.; Napolitano, C.; Rivolta, I.; Sanna, T.; Bellocci, F.; Russo, M.A. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005, 112, 3680–3687. [Google Scholar] [CrossRef]
- Nademanee, K.; Raju, H.; De Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef]
- Pieroni, M.; Notarstefano, P.; Oliva, A.; Campuzano, O.; Santangeli, P.; Coll, M.; Nesti, M.; Carnevali, A.; Fraticelli, A.; Iglesias, A.; et al. Electroanatomic and Pathologic Right Ventricular Outflow Tract Abnormalities in Patients With Brugada Syndrome. J. Am. Coll. Cardiol. 2018, 72, 2747–2757. [Google Scholar] [CrossRef]
- Mitroi, C.; García-Izquierdo, E.; García-Lunar, I.; Castro-Urda, V.; Toquero-Ramos, J.; Moñivas-Palomero, V.; Mingo-Santos, S.; Cavero, M.A.; Brugada, J.; Fernández-Lozano, I. Right ventricular function and dyssynchrony in Brugada syndrome: Highlighting the importance of the mechanical substrate in the right ventricular outflow tract. Int. J. Cardiol. 2021, 333, 233–238. [Google Scholar] [CrossRef]
- Pappone, C.; Santinelli, V.; Mecarocci, V.; Tondi, L.; Ciconte, G.; Manguso, F.; Sturla, F.; Vicedomini, G.; Micaglio, E.; Anastasia, L.; et al. Brugada Syndrome: New Insights From Cardiac Magnetic Resonance and Electroanatomical Imaging. Circ. Arrhythm. Electrophysiol. 2021, 14, E010004. [Google Scholar] [CrossRef]
- Moncayo-Arlandi, J.; Brugada, R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat. Rev. Cardiol. 2017, 14, 744–756. [Google Scholar] [CrossRef]
- Ben-Haim, Y.; Asimaki, A.; Behr, E.R. Brugada syndrome and arrhythmogenic cardiomyopathy: Overlapping disorders of the connexome? Europace 2021, 23, 653–664. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef] [PubMed]
- Saura, D.; García-Alberola, A.; Carrillo, P.; Pascual, D.; Martínez-Sánchez, J.; Valdés, M. Brugada-like electrocardiographic pattern induced by fever. Pacing Clin. Electrophysiol. 2002, 25, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Kawada, S.; Morita, H.; Antzelevitch, C.; Morimoto, Y.; Nakagawa, K.; Watanabe, A.; Nishii, N.; Nakamura, K.; Ito, H. Shanghai Score System for Diagnosis of Brugada Syndrome: Validation of the Score System and System and Reclassification of the Patients. JACC Clin. Electrophysiol. 2018, 4, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.X.; Antzelevitch, C. Cellular basis for the electrocardiographic J wave. Circulation 1996, 93, 372–379. [Google Scholar] [CrossRef]
- Szél, T.; Antzelevitch, C. Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of brugada syndrome. J. Am. Coll. Cardiol. 2014, 63, 2037–2045. [Google Scholar] [CrossRef]
- Behr, E.R.; Ben-Haim, Y.; Ackerman, M.J.; Krahn, A.D.; Wilde, A.A.M. Brugada syndrome and reduced right ventricular outflow tract conduction reserve: A final common pathway? Eur. Heart J. 2021, 42, 1073–1081. [Google Scholar] [CrossRef]
- Gaita, F.; Cerrato, N.; Giustetto, C.; Martino, A.; Bergamasco, L.; Millesimo, M.; Barbonaglia, L.; Carvalho, P.; Caponi, D.; Saglietto, A.; et al. Asymptomatic Patients With Brugada ECG Pattern: Long-Term Prognosis From a Large Prospective Study. Circulation 2023, 148, 1543–1555. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimizu, W.; Ogi, H.; Suenari, K.; Oda, N.; Makita, Y.; Kajihara, K.; Hirai, Y.; Sairaku, A.; Tokuyama, T.; et al. A spontaneous Type 1 electrocardiogram pattern in lead V2 is an independent predictor of ventricular fibrillation in Brugada syndrome. Europace 2010, 12, 410–416. [Google Scholar] [CrossRef]
- Sarkozy, A.; Chierchia, G.B.; Paparella, G.; Boussy, T.; De Asmundis, C.; Roos, M.; Henkens, S.; Kaufman, L.; Buyl, R.; Brugada, R.; et al. Inferior and lateral electrocardiographic repolarization abnormalities in brugada syndrome. Circ. Arrhythm. Electrophysiol. 2009, 2, 154–161. [Google Scholar] [CrossRef]
- Kamakura, S.; Ohe, T.; Nakazawa, K.; Aizawa, Y.; Shimizu, A.; Horie, M.; Ogawa, S.; Okumura, K.; Tsuchihashi, K.; Sugi, K.; et al. Long-term prognosis of probands with brugada-pattern ST-elevation in leads V 1-V 3. Circ. Arrhythm. Electrophysiol. 2009, 2, 495–503. [Google Scholar] [CrossRef]
- Kawata, H.; Morita, H.; Yamada, Y.; Noda, T.; Satomi, K.; Aiba, T.; Isobe, M.; Nagase, S.; Nakamura, K.; Kusano, K.F.; et al. Prognostic significance of early repolarization in inferolateral leads in Brugada patients with documented ventricular fibrillation: A novel risk factor for Brugada syndrome with ventricular fibrillation. Heart Rhythm 2013, 10, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Castro Hevia, J.; Antzelevitch, C.; Tornés Bárzaga, F.; Dorantes Sánchez, M.; Dorticós Balea, F.; Zayas Molina, R.; Quiñones Pérez, M.A.; Fayad Rodríguez, Y. Tpeak-Tend and Tpeak-Tend Dispersion as Risk Factors for Ventricular Tachycardia/Ventricular Fibrillation in Patients With the Brugada Syndrome. J. Am. Coll. Cardiol. 2006, 47, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Nagase, S.; Kataoka, N.; Morita, H.; Kamakura, T.; Ueoka, A.; Nakamura, T.; Oka, S.; Miyazaki, Y.; Wakamiya, A.; Nakajima, K.; et al. Demonstration of Arrhythmia Substrate-Associated Dispersion of Repolarization by Epicardial Unipolar Mapping in Brugada Syndrome. JACC Clin. Electrophysiol. 2024, 10, 1576–1588. [Google Scholar] [CrossRef]
- Tada, T.; Kusano, K.F.; Nagase, S.; Banba, K.; Miura, D.; Nishii, N.; Watanabe, A.; Nakamura, K.; Morita, H.; Ohe, T. Clinical significance of macroscopic T-wave alternans after sodium channel blocker administration in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2008, 19, 56–61. [Google Scholar] [CrossRef]
- Ikeda, T.; Sakurada, H.; Sakabe, K.; Sakata, T.; Takami, M.; Tezuka, N.; Nakae, T.; Noro, M.; Enjoji, Y.; Tejima, T.; et al. Assessment of Noninvasive Markers in Identifying Patients at Risk in the Brugada Syndrome: Insight Into Risk Stratification. J. Am. Coll. Cardiol. 2001, 37, 1628–1634. [Google Scholar] [CrossRef]
- Takagi, M.; Yokoyama, Y.; Aonuma, K.; Aihara, N.; Hiraoka, M. Clinical characteristics and risk stratification in symptomatic and asymptomatic patients with Brugada syndrome: Multicenter study in Japan. J. Cardiovasc. Electrophysiol. 2007, 18, 1244–1251. [Google Scholar] [CrossRef]
- Junttila, M.J.; Brugada, P.; Hong, K.; Lizotte, E.; DEZutter, M.; Sarkozy, A.; Brugada, J.; Benito, B.; Perkiomaki, J.S.; Mäkikallio, T.H.; et al. Differences in 12-lead electrocardiogram between symptomatic and asymptomatic Brugada syndrome patients. J. Cardiovasc. Electrophysiol. 2008, 19, 380–383. [Google Scholar] [CrossRef]
- Morita, H.; Kusano, K.F.; Miura, D.; Nagase, S.; Nakamura, K.; Morita, S.T.; Ohe, T.; Zipes, D.P.; Wu, J. Fragmented QRS as a marker of conduction abnormality and a predictor of prognosis of Brugada syndrome. Circulation 2008, 118, 1697–1704. [Google Scholar] [CrossRef]
- Morita, H.; Watanabe, A.; Kawada, S.; Miyamoto, M.; Morimoto, Y.; Nakagawa, K.; Nishii, N.; Nakamura, K.; Ito, H. Identification of electrocardiographic risk markers for the initial and recurrent episodes of ventricular fibrillation in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2018, 29, 107–114. [Google Scholar] [CrossRef]
- Babai Bigi, M.A.; Aslani, A.; Shahrzad, S. aVR sign as a risk factor for life-threatening arrhythmic events in patients with Brugada syndrome. Heart Rhythm 2007, 4, 1009–1012. [Google Scholar] [CrossRef]
- Calò, L.; Giustetto, C.; Martino, A.; Sciarra, L.; Cerrato, N.; Marziali, M.; Rauzino, J.; Carlino, G.; de Ruvo, E.; Guerra, F.; et al. A New Electrocardiographic Marker of Sudden Death in Brugada Syndrome The S-Wave in Lead I. J. Am. Coll. Cardiol. 2016, 67, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Ragab, A.A.Y.; Houck, C.A.; van der Does, L.J.M.E.; Lanters, E.A.H.; Muskens, A.J.Q.M.; de Groot, N.M.S. Prediction of ventricular tachyarrhythmia in Brugada syndrome by right ventricular outflow tract conduction delay signs. J. Cardiovasc. Electrophysiol. 2018, 29, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Sroubek, J.; Probst, V.; Mazzanti, A.; Delise, P.; Hevia, J.C.; Ohkubo, K.; Zorzi, A.; Champagne, J.; Kostopoulou, A.; Yin, X.; et al. Programmed ventricular stimulation for risk stratification in the Brugada syndrome: A pooled analysis. Circulation. 2016, 133, 622–630. [Google Scholar] [CrossRef]
- Pappone, C.; Ciconte, G.; Manguso, F.; Vicedomini, G.; Mecarocci, V.; Conti, M.; Giannelli, L.; Pozzi, P.; Borrelli, V.; Menicanti, L.; et al. Assessing the Malignant Ventricular Arrhythmic Substrate in Patients With Brugada Syndrome. J. Am. Coll. Cardiol. 2018, 71, 1631–1646. [Google Scholar] [CrossRef]
- Alings, M.; Wilde, A. “Brugada” syndrome: Clinical data and suggested pathophysiological mechanism. Circulation 1999, 99, 666–673. [Google Scholar] [CrossRef]
- Smits, J.P.; Eckardt, L.; Probst, V.; Bezzina, C.R.; Schott, J.J.; Remme, C.A.; Haverkamp, W.; Breithardt, G.; Escande, D.; Schulze-Bahr, E.; et al. Genotype-phenotype relationship in Brugada syndrome: Electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J. Am. Coll. Cardiol. 2002, 40, 350–356. [Google Scholar] [CrossRef]
- Lambiase, P.D.; Ahmed, A.K.; Ciaccio, E.J.; Brugada, R.; Lizotte, E.; Chaubey, S.; Ben-Simon, R.; Chow, A.W.; Lowe, M.D.; McKenna, W.J. High-density substrate mapping in brugada syndrome: Combined role of conduction and repolarization heterogeneities in arrhythmogenesis. Circulation 2009, 120, 106–117. [Google Scholar] [CrossRef]
- Maury, P.; Rollin, A.; Sacher, F.; Gourraud, J.B.; Raczka, F.; Pasquié, J.L.; Duparc, A.; Mondoly, P.; Cardin, C.; Delay, M.; et al. Prevalence and prognostic role of various conduction disturbances in patients with the brugada syndrome. Am. J. Cardiol. 2013, 112, 1384–1389. [Google Scholar] [CrossRef]
- Migliore, F.; Testolina, M.; Zorzi, A.; Bertaglia, E.; Silvano, M.; Leoni, L.; Bellin, A.; Basso, C.; Thiene, G.; Allocca, G.; et al. First-degree atrioventricular block on basal electrocardiogram predicts future arrhythmic events in patients with Brugada syndrome: A long-term follow-up study from the Veneto region of Northeastern Italy. Europace 2019, 21, 322–331. [Google Scholar] [CrossRef]
- Rossi, A.; Giannoni, A.; Nesti, M.; Notarstefano, P.; Castiglione, V.; Solarino, G.; Teresi, L.; Mirizzi, G.; Russo, V.; Panchetti, L.; et al. Prognostic value of right ventricular refractory period heterogeneity in Type-1 Brugada electrocardiographic pattern. Europace 2023, 25, 651–659. [Google Scholar] [CrossRef]
- Priori, S.G.; Gasparini, M.; Napolitano, C.; Della Bella, P.; Ottonelli, A.G.; Sassone, B.; Giordano, U.; Pappone, C.; Mascioli, G.; Rossetti, G.; et al. Risk stratification in brugada syndrome: Results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J. Am. Coll. Cardiol. 2012, 59, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Scheirlynck, E.; Van Malderen, S.; Motoc, A.; Lie, Ø.H.; de Asmundis, C.; Sieira, J.; Chierchia, G.B.; Brugada, P.; Cosyns, B.; Droogmans, S. Contraction alterations in Brugada syndrome; association with life-threatening ventricular arrhythmias. Int. J. Cardiol. 2020, 299, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Pappone, C.; Mecarocci, V.; Manguso, F.; Ciconte, G.; Vicedomini, G.; Sturla, F.; Votta, E.; Mazza, B.; Pozzi, P.; Borrelli, V.; et al. New electromechanical substrate abnormalities in high-risk patients with Brugada syndrome. Heart Rhythm 2020, 17, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Catalano, O.; Antonaci, S.; Moro, G.; Mussida, M.; Frascaroli, M.; Baldi, M.; Cobelli, F.; Baiardi, P.; Nastoli, J.; Bloise, R.; et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur. Heart J. 2009, 30, 2241–2248. [Google Scholar] [CrossRef]
- Van Hoorn, F.; Campian, M.E.; Spijkerboer, A.; Blom, M.T.; Planken, R.N.; van Rossum, A.C.; de Bakker, J.M.; Wilde, A.A.; Groenink, M.; Tan, H.L. Scn5a mutations in brugada syndrome are associated with increased cardiac dimensions and reduced contractility. PLoS ONE 2012, 7, e42037. [Google Scholar] [CrossRef]
- Gray, B.; Gnanappa, G.K.; Bagnall, R.D.; Femia, G.; Yeates, L.; Ingles, J.; Burns, C.; Puranik, R.; Grieve, S.M.; Semsarian, C.; et al. Relations between right ventricular morphology and clinical, electrical and genetic parameters in Brugada Syndrome. PLoS ONE 2018, 13, e0195594. [Google Scholar] [CrossRef]
- Bastiaenen, R.; Cox, A.T.; Castelletti, S.; Wijeyeratne, Y.D.; Colbeck, N.; Pakroo, N.; Ahmed, H.; Bunce, N.; Anderson, L.; Moon, J.C.; et al. Late gadolinium enhancement in Brugada syndrome: A marker for subtle underlying cardiomyopathy? Heart Rhythm 2017, 14, 583–589. [Google Scholar] [CrossRef]
- Isbister, J.C.; Gray, B.; Offen, S.; Yeates, L.; Naoum, C.; Medi, C.; Raju, H.; Semsarian, C.; Puranik, R.; Sy, R.W. Longitudinal assessment of structural phenotype in Brugada syndrome using cardiac magnetic resonance imaging. Heart Rhythm O2 2023, 4, 34–41. [Google Scholar] [CrossRef]
- Kukavica, D.; Trancuccio, A.; Mazzanti, A.; Napolitano, C.; Morini, M.; Pili, G.; Memmi, M.; Gambelli, P.; Bloise, R.; Nastoli, J.; et al. Nonmodifiable Risk Factors Predict Outcomes in Brugada Syndrome. J. Am. Coll. Cardiol. 2024, 84, 2087–2098. [Google Scholar] [CrossRef]
- Nademanee, K.; Chung, F.P.; Sacher, F.; Nogami, A.; Nakagawa, H.; Jiang, C.; Hocini, M.; Behr, E.; Veerakul, G.; Jan Smit, J.; et al. Long-Term Outcomes of Brugada Substrate Ablation: A Report from BRAVO (Brugada Ablation of VF Substrate Ongoing Multicenter Registry). Circulation 2023, 147, 1568–1578. [Google Scholar] [CrossRef]
- Marra, M.P.; Leoni, L.; Bauce, B.; Corbetti, F.; Zorzi, A.; Migliore, F.; Silvano, M.; Rigato, I.; Tona, F.; Tarantini, G.; et al. Imaging study of ventricular scar in arrhythmogenic right ventricular cardiomyopathy comparison of 3d standard electroanatomical voltage mapping and contrast-enhanced cardiac magnetic resonance. Circ. Arrhythm. Electrophysiol. 2012, 5, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Casella, M.; Dello Russo, A.; Bergonti, M.; Catto, V.; Conte, E.; Sommariva, E.; Gasperetti, A.; Vettor, G.; Tundo, F.; Sicuso, R.; et al. Diagnostic Yield of Electroanatomic Voltage Mapping in Guiding Endomyocardial Biopsies. Circulation 2020, 142, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Pappone, C.; Berruezo, A.; Vicedomini, G.; Manguso, F.; Ciconte, G.; Giannelli, L.; Santinelli, V. Brugada Syndrome Phenotype Elimination by Epicardial Substrate Ablation. Circ. Arrhythm. Electrophysiol. 2015, 8, 1373–1381. [Google Scholar] [CrossRef]
- Pappone, C.; Brugada, J.; Vicedomini, G.; Ciconte, G.; Manguso, F.; Saviano, M.; Vitale, R.; Cuko, A.; Giannelli, L.; Calovic, Z.; et al. Electrical Substrate Elimination in 135 Consecutive Patients with Brugada Syndrome. Circ. Arrhythm. Electrophysiol. 2017, 10, e005053. [Google Scholar] [CrossRef]
- Talib, A.K.; Takagi, M.; Shimane, A.; Nakano, M.; Hayashi, T.; Okajima, K.; Kentaro, M.; Fukada, K.; Kowase, S.; Kurosaki, K.; et al. Efficacy of Endocardial Ablation of Drug-Resistant Ventricular Fibrillation in Brugada Syndrome. Circ. Arrhythm. Electrophysiol. 2018, 11, e005631. [Google Scholar] [CrossRef]
- Cheniti, G.; Haissaguerre, M.; Dina, C.; Kamakura, T.; Duchateau, J.; Sacher, F.; Racine, H.P.; Surget, E.; Simonet, F.; Gourraud, J.B.; et al. Left Ventricular Abnormal Substrate in Brugada Syndrome. JACC Clin. Electrophysiol. 2023, 9, 2041–2051. [Google Scholar] [CrossRef]
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.; Verkerk, A.O.; de Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777. [Google Scholar] [CrossRef]
- Santangeli, P.; Hamilton-Craig, C.; Dello Russo, A.; Pieroni, M.; Casella, M.; Pelargonio, G.; Di Biase, L.; Smaldone, C.; Bartoletti, S.; Narducci, M.L.; et al. Imaging of scar in patients with ventricular arrhythmias of right ventricular origin: Cardiac magnetic resonance versus electroanatomic mapping. J. Cardiovasc. Electrophysiol. 2011, 22, 1359–1366. [Google Scholar] [CrossRef]
- Chatterjee, D.; Pieroni, M.; Fatah, M.; Charpentier, F.; Cunningham, K.S.; Spears, D.A.; Chatterjee, D.; Suna, G.; Bos, J.M.; Ackerman, M.J.; et al. An autoantibody profile detects Brugada syndrome and identifies abnormally expressed myocardial proteins. Eur. Heart J. 2020, 41, 2878–2890. [Google Scholar] [CrossRef]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria differential diagnosis. Eur. Heart J. 2020, 41, 1414–1427. [Google Scholar] [CrossRef]
- Del Franco, A.; Ruggieri, R.; Pieroni, M.; Ciabatti, M.; Zocchi, C.; Biagioni, G.; Tavanti, V.; Del Pace, S.; Leone, O.; Favale, S.; et al. Atlas of Regional Left Ventricular Scar in Nonischemic Cardiomyopathies: Substrates and Etiologies. JACC Adv. 2024, 3, 101214. [Google Scholar] [CrossRef] [PubMed]
- Santangeli, P.; Dello Russo, A.; Pieroni, M.; Casella, M.; Di Biase, L.; Burkhardt, J.D.; Sanchez, J.; Lakkireddy, D.; Carbucicchio, C.; Zucchetti, M.; et al. Fragmented and delayed electrograms within fibrofatty scar predict arrhythmic events in arrhythmogenic right ventricular cardiomyopathy: Results from a prospective risk stratification study. Heart Rhythm 2012, 9, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/Dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef]
- Santangeli, P.; Pieroni, M.; Dello Russo, A.; Casella, M.; Pelargonio, G.; Macchione, A.; Camporeale, A.; Smaldone, C.; Bartoletti, S.; Di Biase, L.; et al. Noninvasive diagnosis of electroanatomic abnormalities in arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2010, 3, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Nava, A.; Buja, G.; Martini, B.; Fasoli, G.; Oselladore, L.; Turrini, P.; Thiene, G. Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J. Am. Coll. Cardiol. 1996, 27, 443–448. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Buja, G.; Nava, A.; Rossi, L.; Thiene, G. Right bundle branch block, right precordial st-segment elevation, and sudden death in young people. Circulation 2001, 103, 710–717. [Google Scholar] [CrossRef]
- Letsas, K.P.; Efremidis, M.; Weber, R.; Korantzopoulos, P.; Protonotarios, N.; Prappa, E.; Kounas, S.P.; Evagelidou, E.N.; Xydonas, S.; Kalusche, D.; et al. Epsilon-like waves and ventricular conduction abnormalities in subjects with type 1 ECG pattern of Brugada syndrome. Heart Rhythm 2011, 8, 874–878. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Agullo-Pascual, E.; Cerrone, M.; Delmar, M. Arrhythmogenic cardiomyopathy and Brugada syndrome: Diseases of the connexome. FEBS Lett. 2014, 588, 1322–1330. [Google Scholar] [CrossRef]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gärtner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Meshkov, A.; Myasnikov, R.; Kiseleva, A.; Kulikova, O.; Klauke, B.; Sotnikova, E.; Stanasiuk, C.; Divashuk, M.; Pohl, G.M.; et al. Hemi- and Homozygous Loss-of-Function Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset. Int. J. Mol. Sci. 2021, 22, 3786. [Google Scholar] [CrossRef]
- Iacoviello, M.; Forleo, C.; Puzzovivo, A.; Nalin, I.; Guida, P.; Anaclerio, M.; Marangelli, V.; Sorrentino, S.; Monitillo, F.; Ciccone, M.M.; et al. Altered two-dimensional strain measures of the right ventricle in patients with Brugada syndrome and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur. J. Echocardiogr. 2011, 12, 773–781. [Google Scholar] [CrossRef]
- Heermann, P.; Fritsch, H.; Koopmann, M.; Sporns, P.; Paul, M.; Heindel, W.; Schulze-Bahr, E.; Schülke, C. Biventricular myocardial strain analysis using cardiac magnetic resonance feature tracking (CMR-FT) in patients with distinct types of right ventricular diseases comparing arrhythmogenic right ventricular cardiomyopathy (ARVC), right ventricular outflow-tract tachycardia (RVOT-VT), and Brugada syndrome (BrS). Clinical Research in Cardiology. Clin. Res. Cardiol. 2019, 108, 1147–1162. [Google Scholar] [CrossRef]
- Bariani, R.; Cipriani, A.; Rizzo, S.; Celeghin, R.; Bueno Marinas, M.; Giorgi, B.; De Gaspari, M.; Rigato, I.; Leoni, L.; Zorzi, A.; et al. “Hot phase” clinical presentation in arrhythmogenic cardiomyopathy. Europace 2021, 23, 907–917. [Google Scholar] [CrossRef]
- Peretto, G.; Casella, M.; Merlo, M.; Benedetti, S.; Rizzo, S.; Cappelletto, C.; Di Resta, C.; Compagnucci, P.; De Gaspari, M.; Dello Russo, A.; et al. Inflammation on Endomyocardial Biopsy Predicts Risk of MACE in Undefined Left Ventricular Arrhythmogenic Cardiomyopathy. JACC Clin. Electrophysiol. 2023, 9, 951–961. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence from Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies with Disease Severity and Family History. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef]
- Bassetto, G.; Angriman, F.; Gava, C.P.L.D.; Paldino, A.; Perotto, M.; Bordignon, L.; Gigli, M.; Ferro, M.D.; Massa, L.; Altinier, A.; et al. Hot Phases Cardiomyopathy: Pathophysiology, Diagnostic Challenges, and Emerging Therapies. Curr. Cardiol. Rep. 2025, 27, 11. [Google Scholar] [CrossRef]
- McColl, H.; Cordina, R.; Lal, S.; Parker, M.; Hunyor, I.; Medi, C.; Gray, B. Recurrent immunosuppressive-responsive myocarditis in a patient with desmoplakin cardiomyopathy: A case report. Eur. Heart J. Case Rep. 2024, 8, ytae129. [Google Scholar] [CrossRef]
- Kuroki, K.; Yamamoto, M.; Sato, A. Successful treatment of life-threatening giant cell myocarditis in a patient with Brugada syndrome, controlled through dual ablation procedures and immunosuppressive therapy. Eur. Heart J. Cardiovasc. Imaging 2024, 25, E97. [Google Scholar] [CrossRef]
- Peretto, G.; De Luca, G.; Villatore, A.; Di Resta, C.; Sala, S.; Palmisano, A.; Vignale, D.; Campochiaro, C.; Lazzeroni, D.; De Gaspari, M.; et al. Multimodal Detection and Targeting of Biopsy-Proven Myocardial Inflammation in Genetic Cardiomyopathies: A Pilot Report. JACC Basic Transl. Sci. 2023, 8, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Tung, R.; Shivkumar, K.; Bradfield, J.S. Brugada syndrome—Malignant phenotype associated with acute cardiac inflammation? HeartRhythm Case Rep. 2017, 3, 384–388. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Clinical and Genetic Variables | ECG | Imaging | EPS |
|---|---|---|---|
| Elevates Shanghai score values [23] Family history of SCD [30,41] Unexplained syncope [43,48,49,51,59] Missense SCN5A mutations in BrS-enriched domains [59] Polygenic risk score for BrS [59] | Spontaneous type 1 Brugada pattern [27,28,43,48,49,51,59] LPs on SAE-ECG [35,36] Increased QRS duration [37,38] Fragmented QRS [39,40,43] Tpeak to Tend [33,40] Early repolarization pattern in inferolateral leads [30,31,39] Tall R wave in aVR [40] S-wave duration in lead I ≥ 40 ms [41] Atrial fibrillation [41] Deep S waves in lead I [42] S in lead II > S in lead III [42] Positive Tzou criteria [42] Atrioventricular block [48,49] TWA during sodium channels blocker infusion [34] | Increased RV index of myocardial performance [17] Reduced RVOT values on STE [17] Increased RVMD on STE [17] Increased LVMD on STE [52] RV dysfunction on three-dimensional echocardiography [53] | VT/VF inducibility during EPS [27,41,43] Reduced RV refractory period [51] Heterogenous RV refractory period [50] Persistence of BrS pattern after RVOT ablation [60] |
| Study | Population | Design | Pathological Findings | Notes |
|---|---|---|---|---|
| Coronel et al. [67] | 1 BrS patient undergoing heart transplantation due to refractory electrical storm. | Histopathological and electrophysiological evaluation of the explanted heart. | Presence of fibrous and fatty infiltrations in RV and RVOT. | Presence of conduction delay in RVOT at electrophysiological study. |
| Frustaci et al. [14] | 18 BrS patients suffering arrhythmic events. | Biventricular angiography and EMBs. | Activated T lymphocytes and necrosis in 14/18 patients. Myocyte vacuolization and fibrofatty replacement in 3/18 subjects. | Presence of viral genome in 28% of subjects. Evidence of microaneurysm in the RV and LV (7 and 4 subjects, respectively) at angiography. |
| Nademanee et al. [15] | 6 BrS patients suffering SCD and 6 BrS subjects undergoing surgical epicardial RVOT ablation. | Histopathological evaluation of the explanted heart. RVOT mapping and biopsies in patients undergoing surgical ablation. | Increased fibrosis and collagen in the RVOT. Reduced Cx43 expression in BrS patients. | Presence of delayed, prolonged and fragmented QRS on RVOT epicardial electrocardiogram in patients undergoing ablation. |
| Pieroni et al. [16] | 30 BrS patients (37% of them symptomatic). | 30 BrS patients underwent EPS and endocardial RV EAM. 20/30 subjects underwent EAM-guided EMB. | Activated T lymphocytes infiltrate in 12/20 subjects and myocyte necrosis in 3 of them. Interstitial fibrosis and replacement necrosis in 15/20 patients. | Patients with inflammation at EMB presented higher rates of VF inducibility and more extensive bipolar abnormalities at EAM. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciabatti, M.; Notarstefano, P.; Zocchi, C.; Virgili, G.; Bellocci, F.; Olivotto, I.; Pieroni, M. Brugada Syndrome: Channelopathy and/or Cardiomyopathy. Cardiogenetics 2025, 15, 17. https://doi.org/10.3390/cardiogenetics15020017
Ciabatti M, Notarstefano P, Zocchi C, Virgili G, Bellocci F, Olivotto I, Pieroni M. Brugada Syndrome: Channelopathy and/or Cardiomyopathy. Cardiogenetics. 2025; 15(2):17. https://doi.org/10.3390/cardiogenetics15020017
Chicago/Turabian StyleCiabatti, Michele, Pasquale Notarstefano, Chiara Zocchi, Giacomo Virgili, Fulvio Bellocci, Iacopo Olivotto, and Maurizio Pieroni. 2025. "Brugada Syndrome: Channelopathy and/or Cardiomyopathy" Cardiogenetics 15, no. 2: 17. https://doi.org/10.3390/cardiogenetics15020017
APA StyleCiabatti, M., Notarstefano, P., Zocchi, C., Virgili, G., Bellocci, F., Olivotto, I., & Pieroni, M. (2025). Brugada Syndrome: Channelopathy and/or Cardiomyopathy. Cardiogenetics, 15(2), 17. https://doi.org/10.3390/cardiogenetics15020017