
Butyrate Improves Neuroinflammation and Mitochondrial Impairment in Cerebral Cortex and Synaptic Fraction in an Animal Model of Diet-Induced Obesity



 ,
,  , , ,
, , ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal Diet
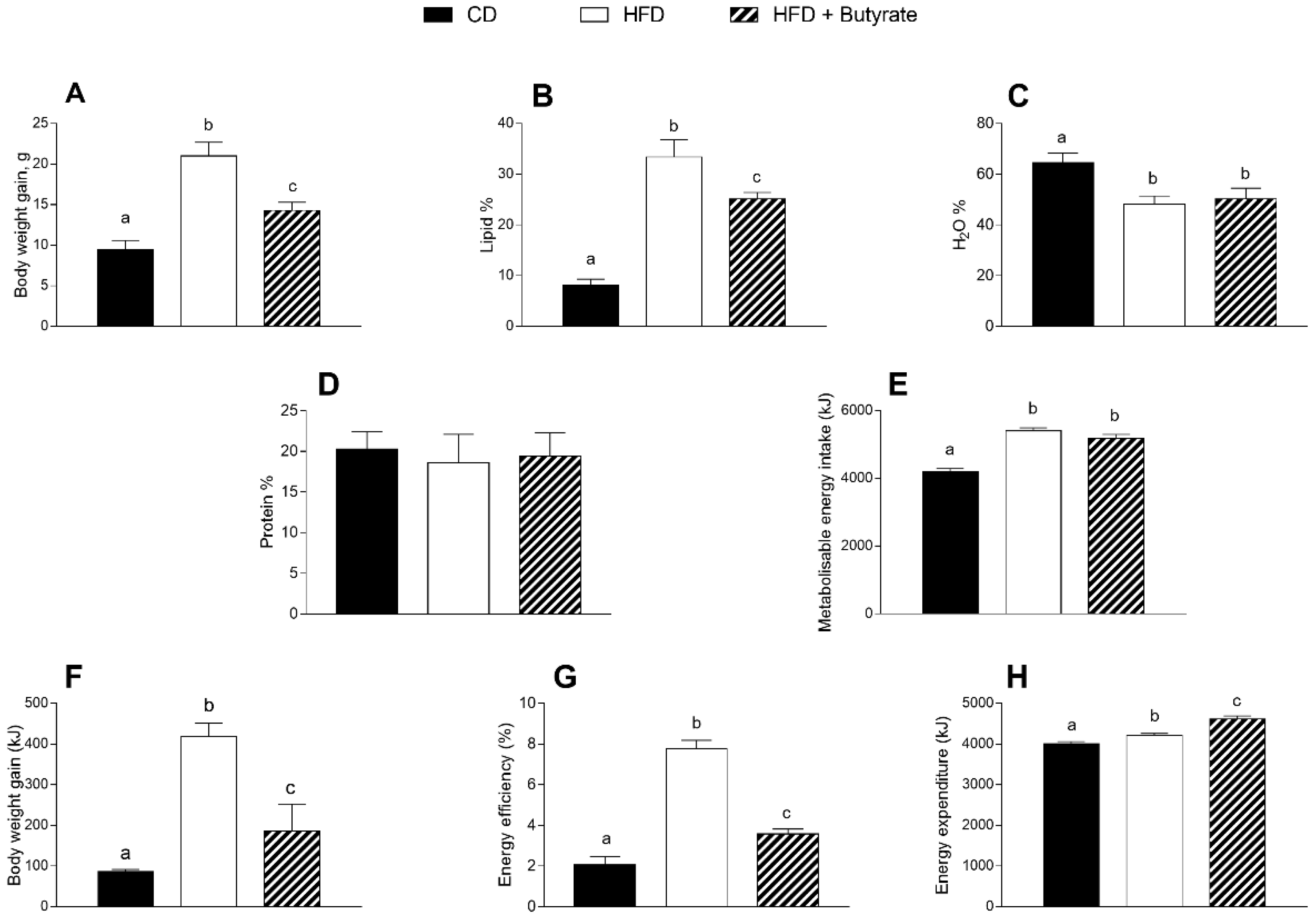
2.3. Body Composition and Energy Balance
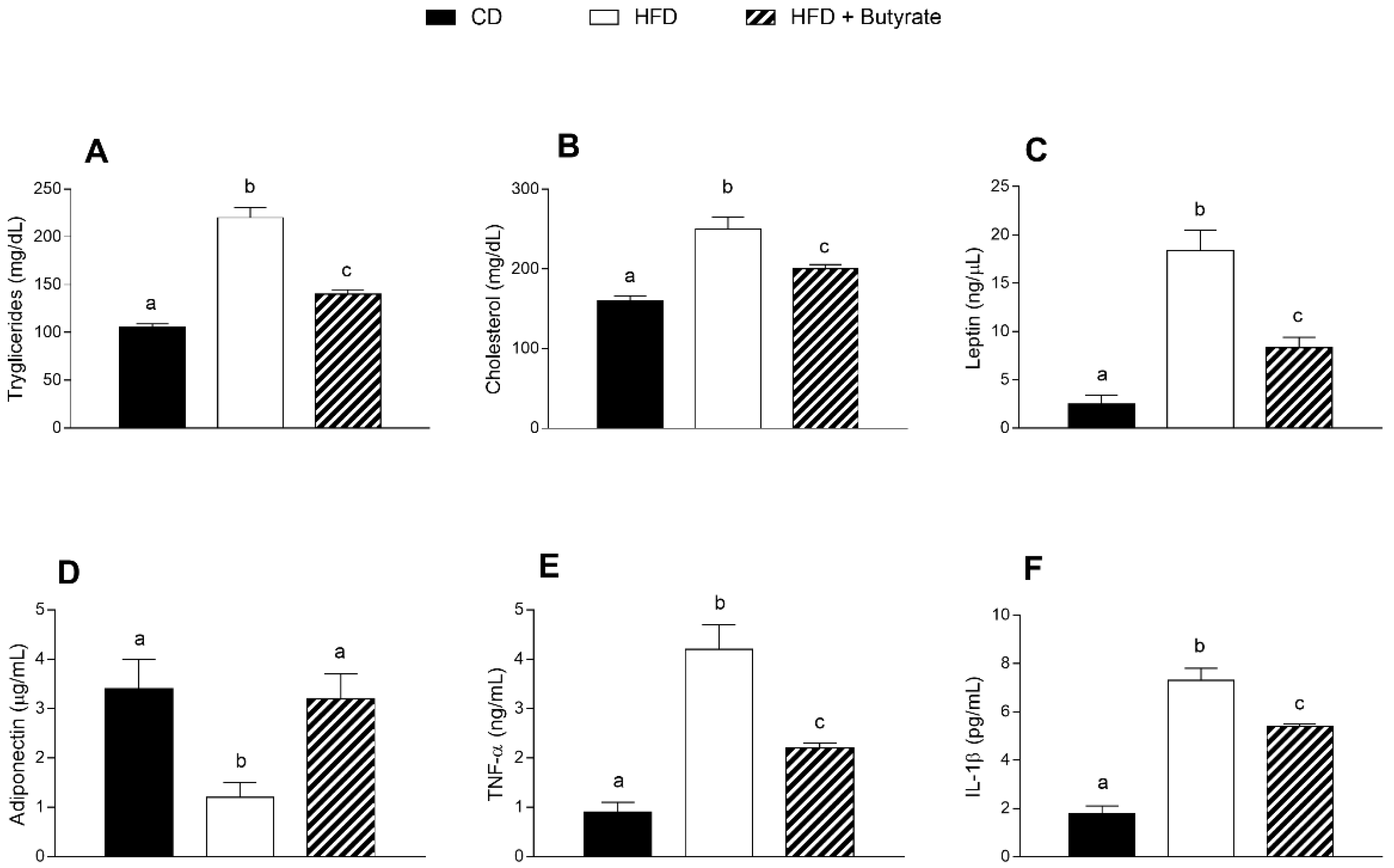
2.4. Serum Parameters
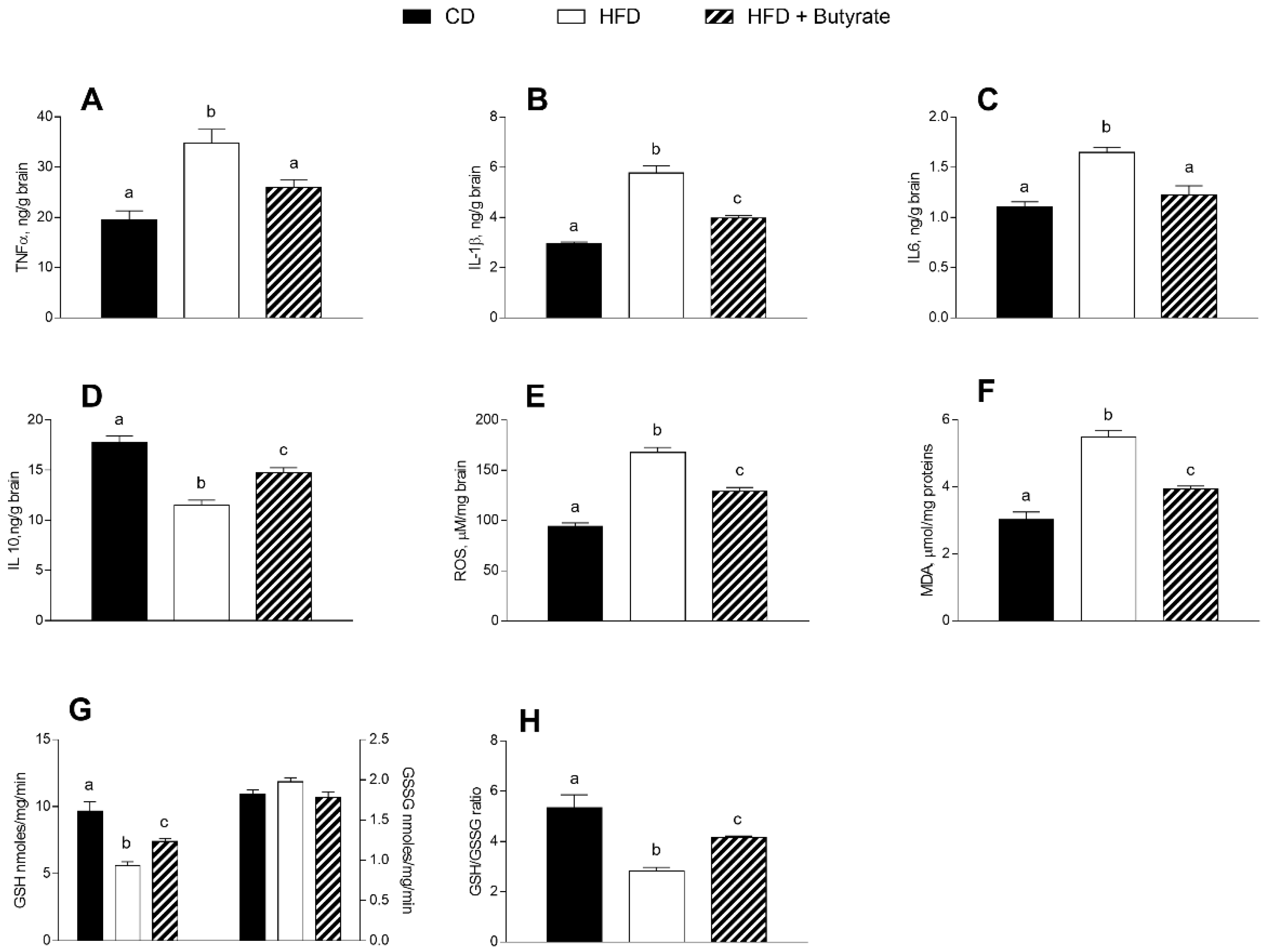
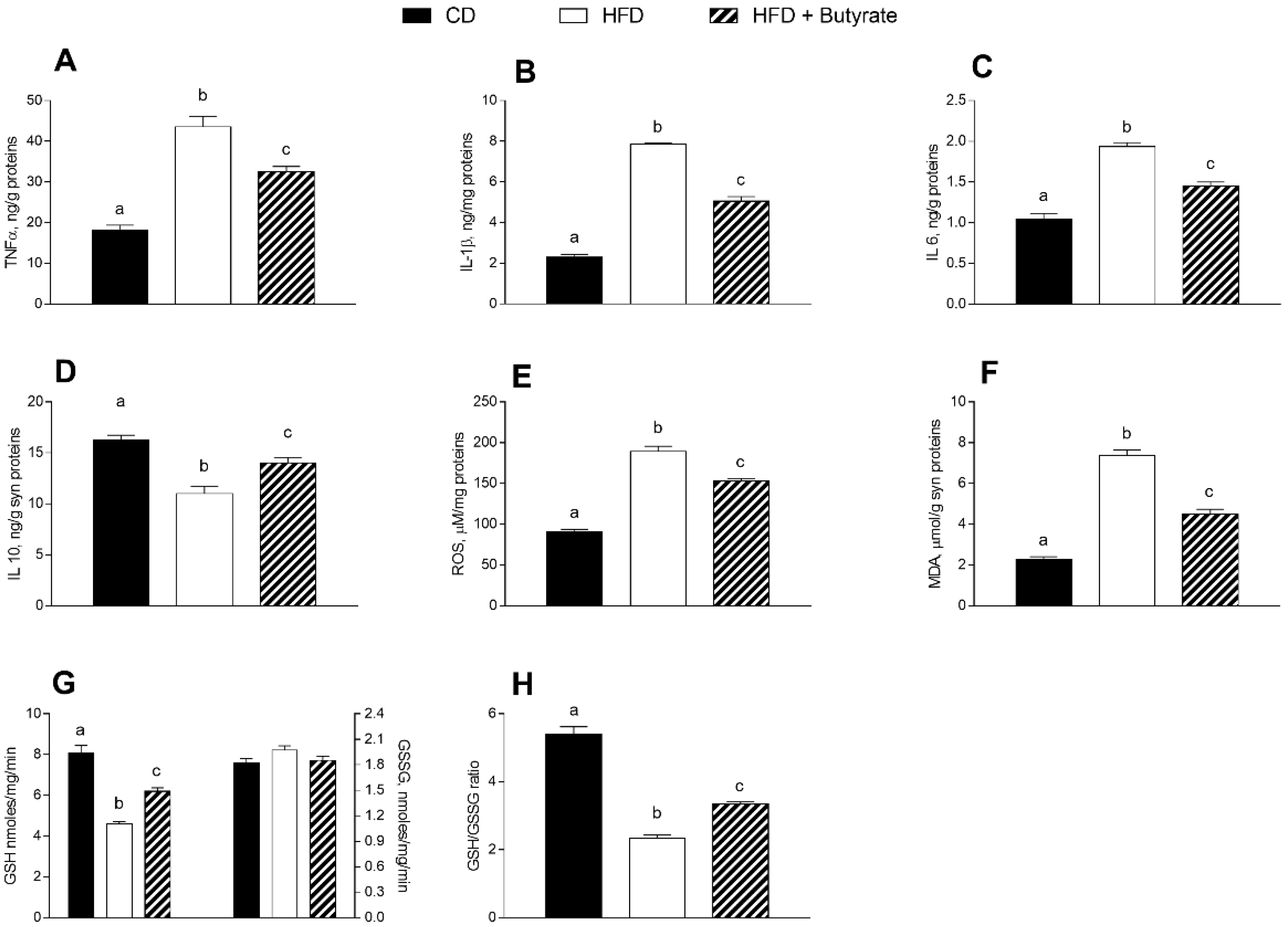
2.5. Brain and Synaptosomes Parameters
2.6. Preparation of Mitochondria and Synaptosomes from the Cerebral Cortex
2.7. Measurements of Mitochondrial Oxidative Capacities and Degree of Coupling
2.8. Seahorse XFp Analyzer Cell Mito Stress Test
2.9. Western Blot Analysis
2.10. Statistical Analysis
3. Results
3.1. Effect of Butyrate on Body Composition and Energy Balance of HFD Mice
3.2. Effect of Butyrate on Serum Metabolic Parameters and Inflammatory Markers of HFD Mice
3.3. Effect of Butyrate on Inflammation and Oxidative Stress in the Brain Cortex of HFD Mice
3.4. Effect of Butyrate on Inflammation and Oxidative Stress in the Synaptic Fraction of HFD Mice
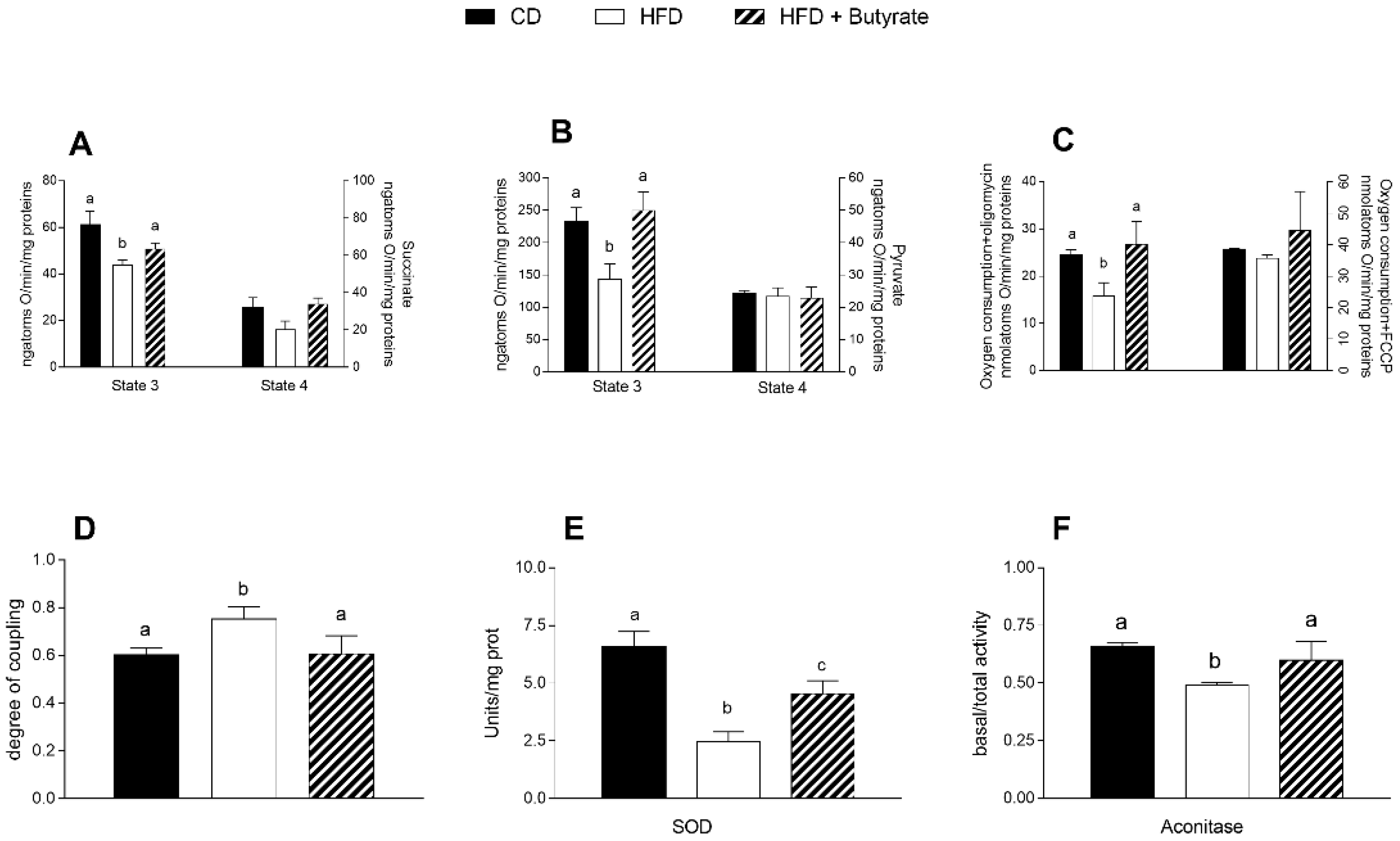
3.5. Effect of Butyrate on Mitochondrial Function, Efficiency and Oxidative Stress in HFD Mouse Brain Cortex
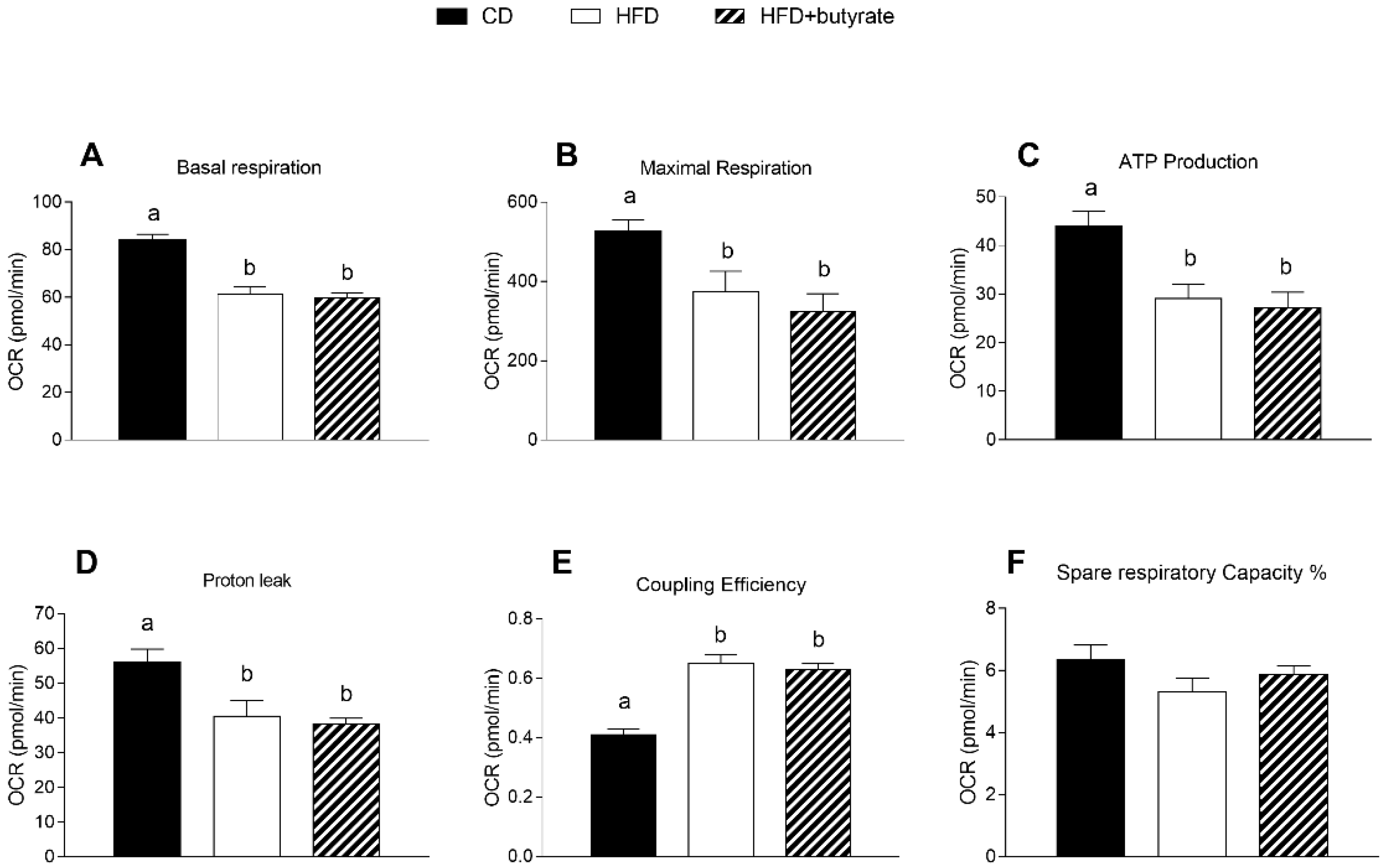
3.6. Effect of Butyrate on Mitochondrial Function in the Synaptic Fraction from HFD Mouse Brain Cortex
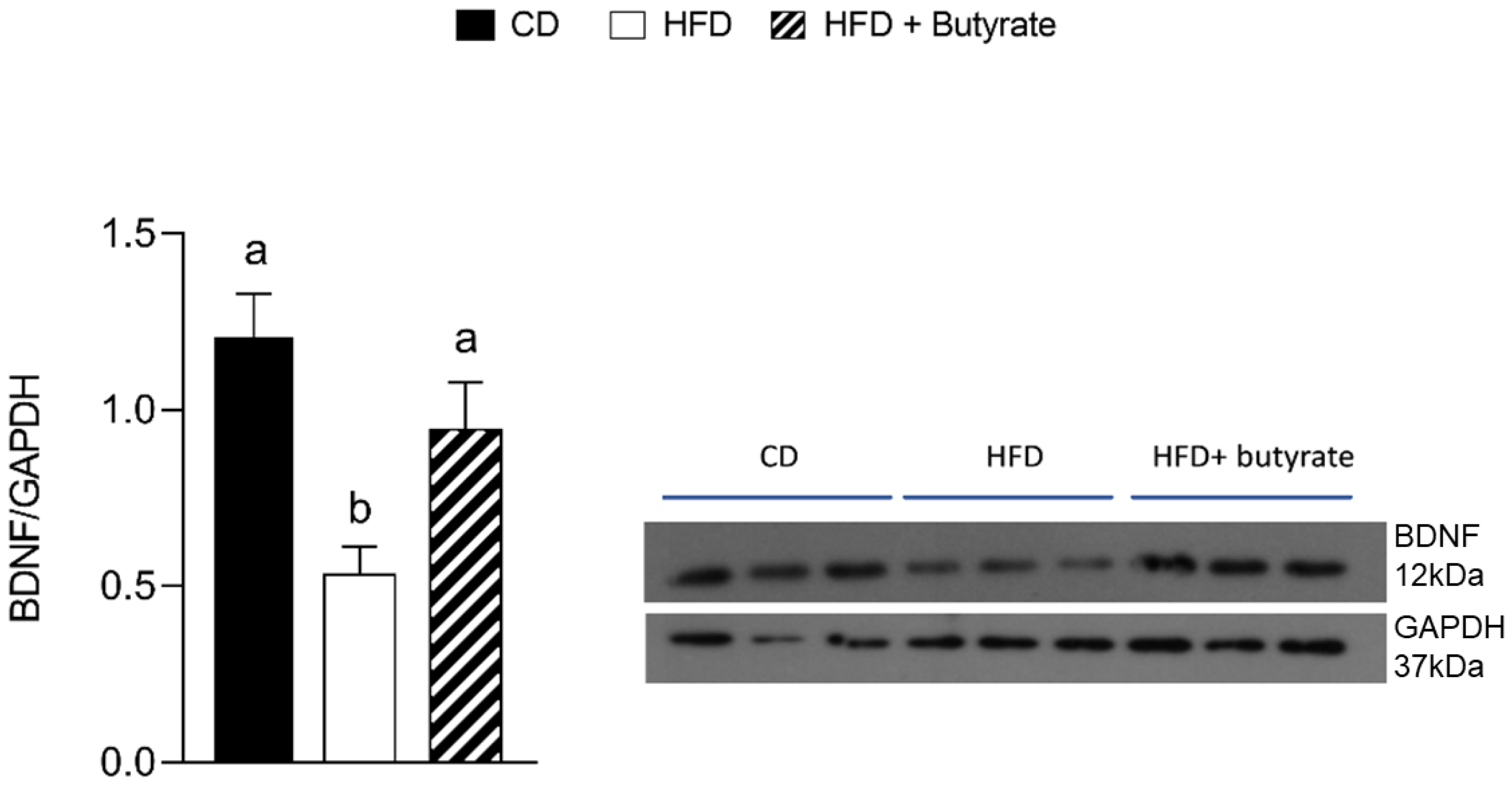
3.7. Effect of Butyrate on BDNF Pathways in the Brain Cortex from HFD Mouse
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agha, M.; Agha, R. The Rising Prevalence of Obesity: Part A: Impact on Public Health. Int. J. Surg. Oncol. 2017, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Azevedo, I. Chronic Inflammation in Obesity and the Metabolic Syndrome. Mediat. Inflamm. 2010, 2010, 289645. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; de Filippo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long Feeding High-Fat Diet Induces Hypothalamic Oxidative Stress and Inflammation, and Prolonged Hypothalamic AMPK Activation in Rat Animal Model. Front. Physiol. 2018, 9, 818. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Trinchese, G.; Bergamo, P.; de Filippo, C.; Mattace Raso, G.; Gifuni, G.; Putti, R.; Moni, B.H.; Canani, R.B.; Meli, R.; et al. Polyunsaturated Fatty Acids Attenuate Diet Induced Obesity and Insulin Resistance, Modulating Mitochondrial Respiratory Uncoupling in Rat Skeletal Muscle. PLoS ONE 2016, 11, e0149033. [Google Scholar] [CrossRef] [PubMed]
- de A. Boleti, A.; de O. Cardoso, P.; Frihling, B.F.; e Silva, P.; de Moraes, L.R.N.; Migliolo, L. Adipose Tissue, Systematic Inflammation, and Neurodegenerative Diseases. Neural Regen. Res. 2023, 18, 38. [Google Scholar] [CrossRef]
- Park, K.-Y.; Nam, G.E.; Han, K.; Park, H.-K.; Hwang, H.-S. Waist Circumference and Risk of Parkinson’s Disease. NPJ Park. Dis. 2022, 8, 89. [Google Scholar] [CrossRef]
- Crispino, M.; Trinchese, G.; Penna, E.; Cimmino, F.; Catapano, A.; Villano, I.; Perrone-Capano, C.; Mollica, M.P. Interplay between Peripheral and Central Inflammation in Obesity-Promoted Disorders: The Impact on Synaptic Mitochondrial Functions. Int. J. Mol. Sci. 2020, 21, 5964. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Dumitrascu, D.; Capitanescu, B.; Petcu, E.; Surugiu, R.; Fang, W.-H.; Dumbrava, D.-A. Dietary Habits, Lifestyle Factors and Neurodegenerative Diseases. Neural Regen. Res. 2020, 15, 394. [Google Scholar] [CrossRef]
- Dai, L.; Zou, L.; Meng, L.; Qiang, G.; Yan, M.; Zhang, Z. Cholesterol Metabolism in Neurodegenerative Diseases: Molecular Mechanisms and Therapeutic Targets. Mol. Neurobiol. 2021, 58, 2183–2201. [Google Scholar] [CrossRef]
- Sabia, S.; Kivimaki, M.; Shipley, M.J.; Marmot, M.G.; Singh-Manoux, A. Body Mass Index over the Adult Life Course and Cognition in Late Midlife: The Whitehall II Cohort Study. Am. J. Clin. Nutr. 2009, 89, 601–607. [Google Scholar] [CrossRef]
- Kronschnabl, J.M.; Kneip, T.; Weiss, L.M.; Bergmann, M. Bodyweight Change and Cognitive Performance in the Older Population. PLoS ONE 2021, 16, e0249651. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ajnakina, O.; Steptoe, A.; Cadar, D. Higher Risk of Dementia in English Older Individuals Who Are Overweight or Obese. Int. J. Epidemiol. 2020, 49, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Lima Giacobbo, B.; Doorduin, J.; Klein, H.C.; Dierckx, R.A.J.O.; Bromberg, E.; de Vries, E.F.J. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Qi, G.; Fan, R.; Qiao, Q.; Sun, Y.; Gao, Y.; Liu, X. EGCG Ameliorates High-fat– and High-fructose-induced Cognitive Defects by Regulating the IRS/AKT and ERK/CREB/BDNF Signaling Pathways in the CNS. FASEB J. 2017, 31, 4998–5011. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yao, M.; Wei, L.; Ge, S. Obesity Caused by a High-Fat Diet Regulates the Sirt1/PGC-1α/FNDC5/BDNF Pathway to Exacerbate Isoflurane-Induced Postoperative Cognitive Dysfunction in Older Mice. Nutr. Neurosci. 2020, 23, 971–982. [Google Scholar] [CrossRef]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef]
- VanGuilder, H.D.; Farley, J.A.; Yan, H.; van Kirk, C.A.; Mitschelen, M.; Sonntag, W.E.; Freeman, W.M. Hippocampal Dysregulation of Synaptic Plasticity-Associated Proteins with Age-Related Cognitive Decline. Neurobiol. Dis. 2011, 43, 201–212. [Google Scholar] [CrossRef]
- Penna, E.; Pizzella, A.; Cimmino, F.; Trinchese, G.; Cavaliere, G.; Catapano, A.; Allocca, I.; Chun, J.T.; Campanozzi, A.; Messina, G.; et al. Neurodevelopmental Disorders: Effect of High-Fat Diet on Synaptic Plasticity and Mitochondrial Functions. Brain Sci. 2020, 10, 805. [Google Scholar] [CrossRef]
- Canani, R.B. Potential Beneficial Effects of Butyrate in Intestinal and Extraintestinal Diseases. World J. Gastroenterol. 2011, 17, 1519. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, K.; Li, X.; Xu, L.; Yang, Z. Sodium Butyrate Ameliorates the Impairment of Synaptic Plasticity by Inhibiting the Neuroinflammation in 5XFAD Mice. Chem. Biol. Interact. 2021, 341, 109452. [Google Scholar] [CrossRef]
- Hou, Y.; Li, X.; Liu, C.; Zhang, M.; Zhang, X.; Ge, S.; Zhao, L. Neuroprotective Effects of Short-Chain Fatty Acids in MPTP Induced Mice Model of Parkinson’s Disease. Exp. Gerontol. 2021, 150, 111376. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, C.; Coretti, L.; Lama, A.; Pirozzi, C.; de Caro, C.; de Biase, D.; Turco, L.; Mollica, M.P.; Paciello, O.; Calignano, A.; et al. Dual-Hit Model of Parkinson’s Disease: Impact of Dysbiosis on 6-Hydroxydopamine-Insulted Mice—Neuroprotective and Anti-Inflammatory Effects of Butyrate. Int. J. Mol. Sci. 2022, 23, 6367. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; de Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef]
- Pirozzi, C.; Lama, A.; Annunziata, C.; Cavaliere, G.; de Caro, C.; Citraro, R.; Russo, E.; Tallarico, M.; Iannone, M.; Ferrante, M.C.; et al. Butyrate Prevents Valproate-induced Liver Injury: In Vitro and in Vivo Evidence. FASEB J. 2020, 34, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Gu, J.; Zhang, H.; Wang, Z.; Zhang, W.; Zhao, Y.; Zheng, Y.; Zhang, W.; Zhou, H.; Zhang, G.; et al. Sodium Butyrate-Modulated Mitochondrial Function in High-Insulin Induced HepG2 Cell Dysfunction. Oxid. Med. Cell. Longev. 2020, 2020, 1904609. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bai, X.; Jiang, Y.; Cheng, Y. Butyrate Alleviates PTZ-Induced Mitochondrial Dysfunction, Oxidative Stress and Neuron Apoptosis in Mice via Keap1/Nrf2/HO-1 Pathway. Brain Res. Bull. 2021, 168, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Stanley, G.H.S. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Brooks, S.P.J.; Lampi, B.J.; Sarwar, G.; Botting, H.G. A Comparison of Methods for Determining Total Body Protein. Anal. Biochem. 1995, 226, 26–30. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, D.-M.; Chen, H.-L.; Lin, Y.-X.; Hang, C.-H.; Yin, H.-X.; Shi, J.-X. N-Acetylcysteine Suppresses Oxidative Stress in Experimental Rats with Subarachnoid Hemorrhage. J. Clin. Neurosci. 2009, 16, 684–688. [Google Scholar] [CrossRef]
- Montoliu, C.; Vallés, S.; Renau-Piqueras, J.; Guerri, C. Ethanol-Induced Oxygen Radical Formation and Lipid Peroxidation in Rat Brain: Effect of Chronic Alcohol Consumption. J. Neurochem. 2002, 63, 1855–1862. [Google Scholar] [CrossRef]
- Viggiano, E.; Mollica, M.P.; Lionetti, L.; Cavaliere, G.; Trinchese, G.; de Filippo, C.; Chieffi, S.; Gaita, M.; Barletta, A.; de Luca, B.; et al. Effects of an High-Fat Diet Enriched in Lard or in Fish Oil on the Hypothalamic Amp-Activated Protein Kinase and Inflammatory Mediators. Front. Cell. Neurosci. 2016, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Steward, O. Evidence That Protein Constituents of Postsynaptic Membrane Specializations Are Locally Synthesized: Analysis of Proteins Synthesized within Synaptosomes. J. Neurosci. 1991, 11, 2881–2895. [Google Scholar] [CrossRef] [PubMed]
- Eyman, M.; Cefaliello, C.; Ferrara, E.; de Stefano, R.; Crispino, M.; Giuditta, A. Synaptosomal Protein Synthesis Is Selectively Modulated by Learning. Brain Res. 2007, 1132, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Eyman, M.; Cefaliello, C.; Mandile, P.; Piscopo, S.; Crispino, M.; Giuditta, A. Training Old Rats Selectively Modulates Synaptosomal Protein Synthesis. J. Neurosci. Res. 2012, 91, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Cefaliello, C.; Eyman, M.; Melck, D.; de Stefano, R.; Ferrara, E.; Crispino, M.; Giuditta, A. Brain Synaptosomes Harbor More than One Cytoplasmic System of Protein Synthesis. J. Neurosci. Res. 2014, 92, 1573–1580. [Google Scholar] [CrossRef]
- Penna, E.; Cerciello, A.; Chambery, A.; Russo, R.; Cernilogar, F.M.; Pedone, E.M.; Perrone-Capano, C.; Cappello, S.; di Giaimo, R.; Crispino, M. Cystatin B Involvement in Synapse Physiology of Rodent Brains and Human Cerebral Organoids. Front. Mol. Neurosci. 2019, 12, 195. [Google Scholar] [CrossRef]
- Estabrook, R.W. [7] Mitochondrial Respiratory Control and the Polarographic Measurement of ADP: O Ratios. Methods Enzymol. 1967, 10, 41–47. [Google Scholar]
- Cairns, C.B.; Walther, J.; Harken, A.H.; Banerjee, A. Mitochondrial Oxidative Phosphorylation Thermodynamic Efficiencies Reflect Physiological Organ Roles. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1998, 274, R1376–R1383. [Google Scholar] [CrossRef]
- Flohé, L.; Otting, F. Superoxide Dismutase Assays. Methods Enzym. 1984, 105, 93–104. [Google Scholar] [CrossRef]
- Chun, J.T.; Crispino, M.; Tocco, G. The Dual Response of Protein Kinase Fyn to Neural Trauma: Early Induction in Neurons and Delayed Induction in Reactive Astrocytes. Exp. Neurol. 2004, 185, 109–119. [Google Scholar] [CrossRef]
- Cristiano, C.; Cuozzo, M.; Coretti, L.; Liguori, F.M.; Cimmino, F.; Turco, L.; Avagliano, C.; Aviello, G.; Mollica, M.P.; Lembo, F.; et al. Oral Sodium Butyrate Supplementation Ameliorates Paclitaxel-Induced Behavioral and Intestinal Dysfunction. Biomed. Pharmacother. 2022, 153, 113528. [Google Scholar] [CrossRef] [PubMed]
- Conte, C.; Sichetti, M.; Traina, G. Gut–Brain Axis: Focus on Neurodegeneration and Mast Cells. Appl. Sci. 2020, 10, 1828. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin Stimulates Glucose Utilization and Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin Stimulates Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, C.K. Role of Adiponectin and Some Other Factors Linking Type 2 Diabetes Mellitus and Obesity. World J. Diabetes 2015, 6, 1296–1308. [Google Scholar] [CrossRef]
- Kim, H.J.; Leeds, P.; Chuang, D.-M. The HDAC Inhibitor, Sodium Butyrate, Stimulates Neurogenesis in the Ischemic Brain. J. Neurochem. 2009, 110, 1226–1240. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Zhang, B.; Tian, J.; Yin, X.; Luo, W.; Xia, K. Protective Effect of Sodium Butyrate on the Cell Culture Model of Huntington Disease. Prog. Nat. Sci. 2017, 17, 784–788. [Google Scholar]
- Bayazid, A.B.; Jang, Y.A.; Kim, Y.M.; Kim, J.G.; Lim, B.O. Neuroprotective Effects of Sodium Butyrate through Suppressing Neuroinflammation and Modulating Antioxidant Enzymes. Neurochem. Res. 2021, 46, 2348–2358. [Google Scholar] [CrossRef]
- Matt, S.M.; Allen, J.M.; Lawson, M.A.; Mailing, L.J.; Woods, J.A.; Johnson, R.W. Butyrate and Dietary Soluble Fiber Improve Neuroinflammation Associated with Aging in Mice. Front. Immunol. 2018, 9, 1832. [Google Scholar] [CrossRef]
- Cefaliello, C.; Penna, E.; Barbato, C.; di Ruberto, G.; Mollica, M.P.; Trinchese, G.; Cigliano, L.; Borsello, T.; Chun, J.T.; Giuditta, A.; et al. Deregulated Local Protein Synthesis in the Brain Synaptosomes of a Mouse Model for Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The Role of Pathological Tau in Synaptic Dysfunction in Alzheimer’s Diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Crispino, M.; Cefaliello, C.; Kaplan, B.; Giuditta, A. Protein Synthesis in Nerve Terminals and the Glia-Neuron Unit. Results Probl. Cell Differ. 2009, 48, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Crispino, M.; Chun, J.T.; Cefaliello, C.; Perrone Capano, C.; Giuditta, A. Local Gene Expression in Nerve Endings. Dev. Neurobiol. 2014, 74, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Jhou, J.-F.; Tai, H.-C. The Study of Postmortem Human Synaptosomes for Understanding Alzheimer’s Disease and Other Neurological Disorders: A Review. Neurol. Ther. 2017, 6, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative Imaging of Energy Expenditure in Human Brain. Neuroimage 2012, 60, 2107–2117. [Google Scholar] [CrossRef]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated Energy Budgets for Neural Computation in the Neocortex and Cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of Reactive Oxygen Species Generation by Mitochondria Oxidizing Different Substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial Dysfunction and the Inflammatory Response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Nedergaard, J.; Ricquier, D.; Kozak, L.P. Uncoupling Proteins: Current Status and Therapeutic Prospects. EMBO Rep. 2005, 6, 917–921. [Google Scholar] [CrossRef]
- Tseng, Y.-H.; Cypess, A.M.; Kahn, C.R. Cellular Bioenergetics as a Target for Obesity Therapy. Nat. Rev. Drug Discov. 2010, 9, 465–482. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Uncoupling: New Approaches to an Old Problem of Bioenergetics. Biochim. Biophys. Acta (BBA)—Bioenerg. 1998, 1363, 100–124. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High Protonic Potential Actuates a Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R. Inhibition of Histone Deacetylase Activity by Butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef]
- Bayazid, A.B.; Kim, J.G.; Azam, S.; Jeong, S.A.; Kim, D.H.; Park, C.W.; Lim, B.O. Sodium Butyrate Ameliorates Neurotoxicity and Exerts Anti-Inflammatory Effects in High Fat Diet-Fed Mice. Food Chem. Toxicol. 2022, 159, 112743. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavaliere, G.; Catapano, A.; Trinchese, G.; Cimmino, F.; Penna, E.; Pizzella, A.; Cristiano, C.; Lama, A.; Crispino, M.; Mollica, M.P. Butyrate Improves Neuroinflammation and Mitochondrial Impairment in Cerebral Cortex and Synaptic Fraction in an Animal Model of Diet-Induced Obesity. Antioxidants 2023, 12, 4. https://doi.org/10.3390/antiox12010004
Cavaliere G, Catapano A, Trinchese G, Cimmino F, Penna E, Pizzella A, Cristiano C, Lama A, Crispino M, Mollica MP. Butyrate Improves Neuroinflammation and Mitochondrial Impairment in Cerebral Cortex and Synaptic Fraction in an Animal Model of Diet-Induced Obesity. Antioxidants. 2023; 12(1):4. https://doi.org/10.3390/antiox12010004
Chicago/Turabian StyleCavaliere, Gina, Angela Catapano, Giovanna Trinchese, Fabiano Cimmino, Eduardo Penna, Amelia Pizzella, Claudia Cristiano, Adriano Lama, Marianna Crispino, and Maria Pina Mollica. 2023. "Butyrate Improves Neuroinflammation and Mitochondrial Impairment in Cerebral Cortex and Synaptic Fraction in an Animal Model of Diet-Induced Obesity" Antioxidants 12, no. 1: 4. https://doi.org/10.3390/antiox12010004
APA StyleCavaliere, G., Catapano, A., Trinchese, G., Cimmino, F., Penna, E., Pizzella, A., Cristiano, C., Lama, A., Crispino, M., & Mollica, M. P. (2023). Butyrate Improves Neuroinflammation and Mitochondrial Impairment in Cerebral Cortex and Synaptic Fraction in an Animal Model of Diet-Induced Obesity. Antioxidants, 12(1), 4. https://doi.org/10.3390/antiox12010004