A Chitosan—Based Liposome Formulation Enhances the In Vitro Wound Healing Efficacy of Substance P Neuropeptide

 ,
, 
Abstract
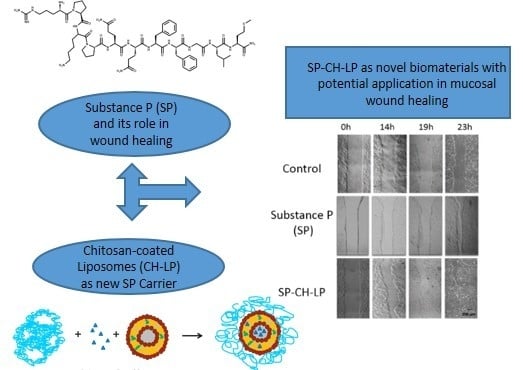
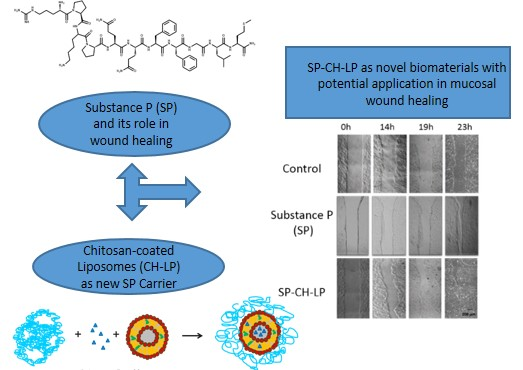
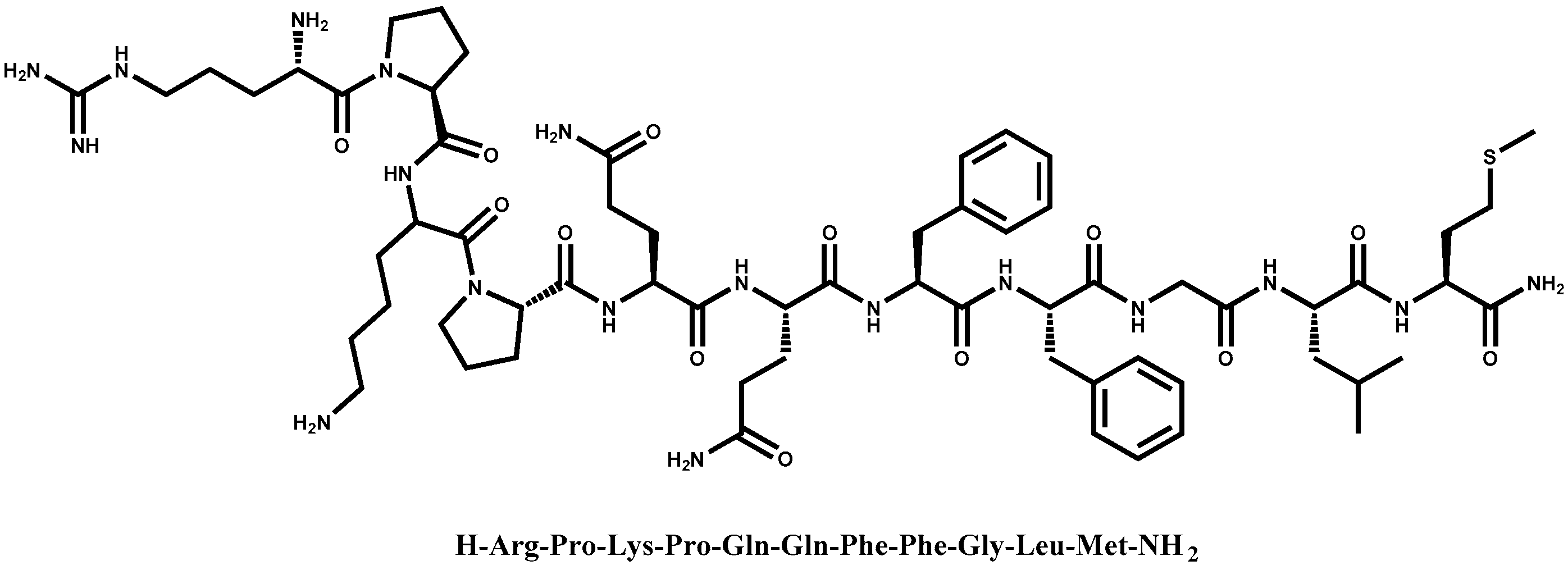
1. Introduction
2. Materials and Methods
2.1. Preparation of Liposomes
2.2. Coating of Liposomes
2.3. Characterization of Chitosan-Coated Liposomes and Uncoated Liposomes
2.3.1. Size and Zeta Potential Measurements
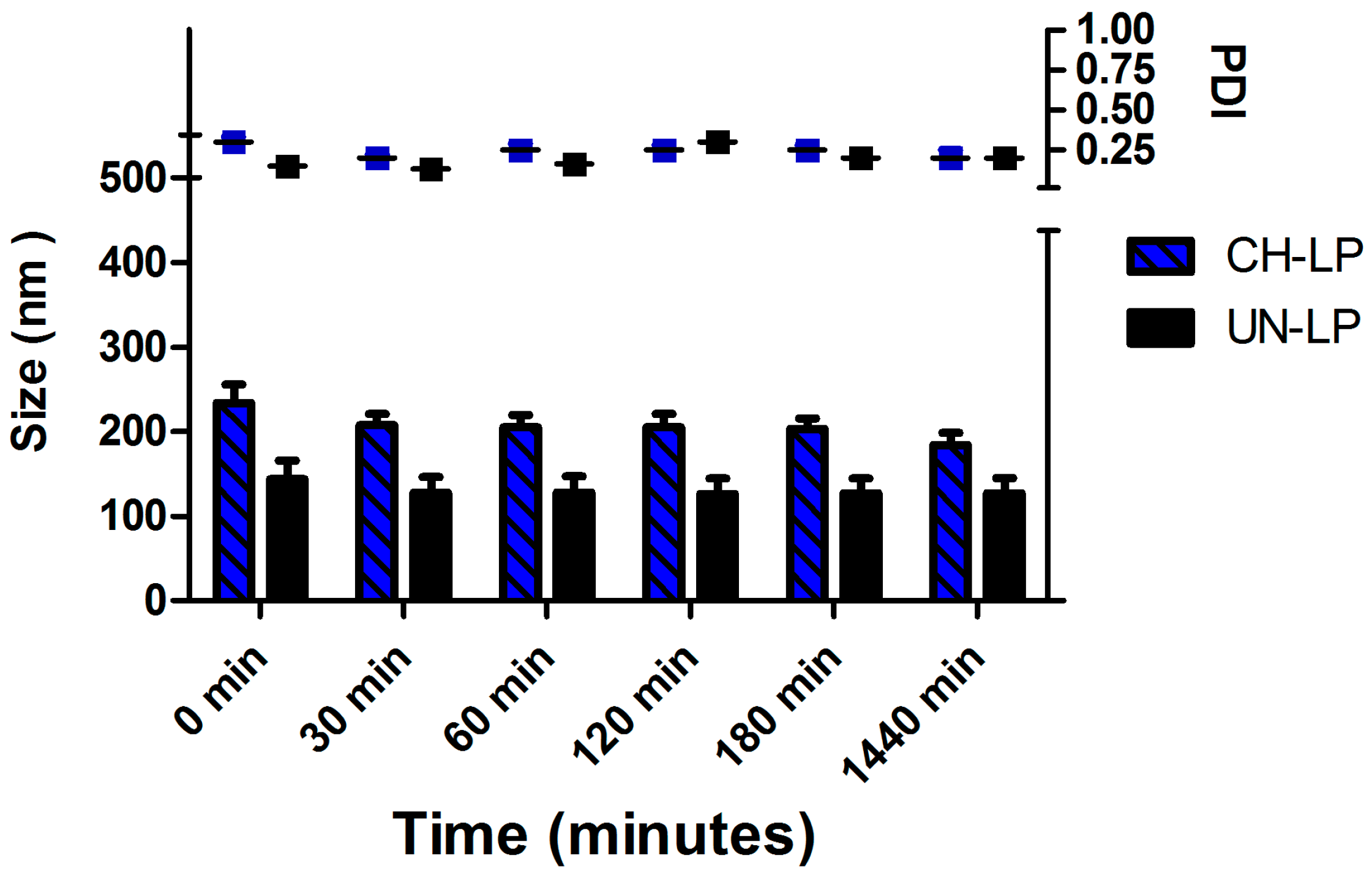
2.3.2. Stability in Physiological Medium
2.3.3. High Performance Liquid Chromatography with Ultraviolet/Light Detection (HPLC/UV-VIS) Analysis
2.3.4. Encapsulation Efficiency
2.3.5. Release Studies
2.4. Cell Culture
2.4.1. Cell Viability Assay
2.4.2. Wound Healing Assay
2.4.3. BrdU Colorimetric Assay
2.4.4. Statistical Analysis
3. Results and Discussion
3.1. Preparation and Characterization of Liposomal Formulations
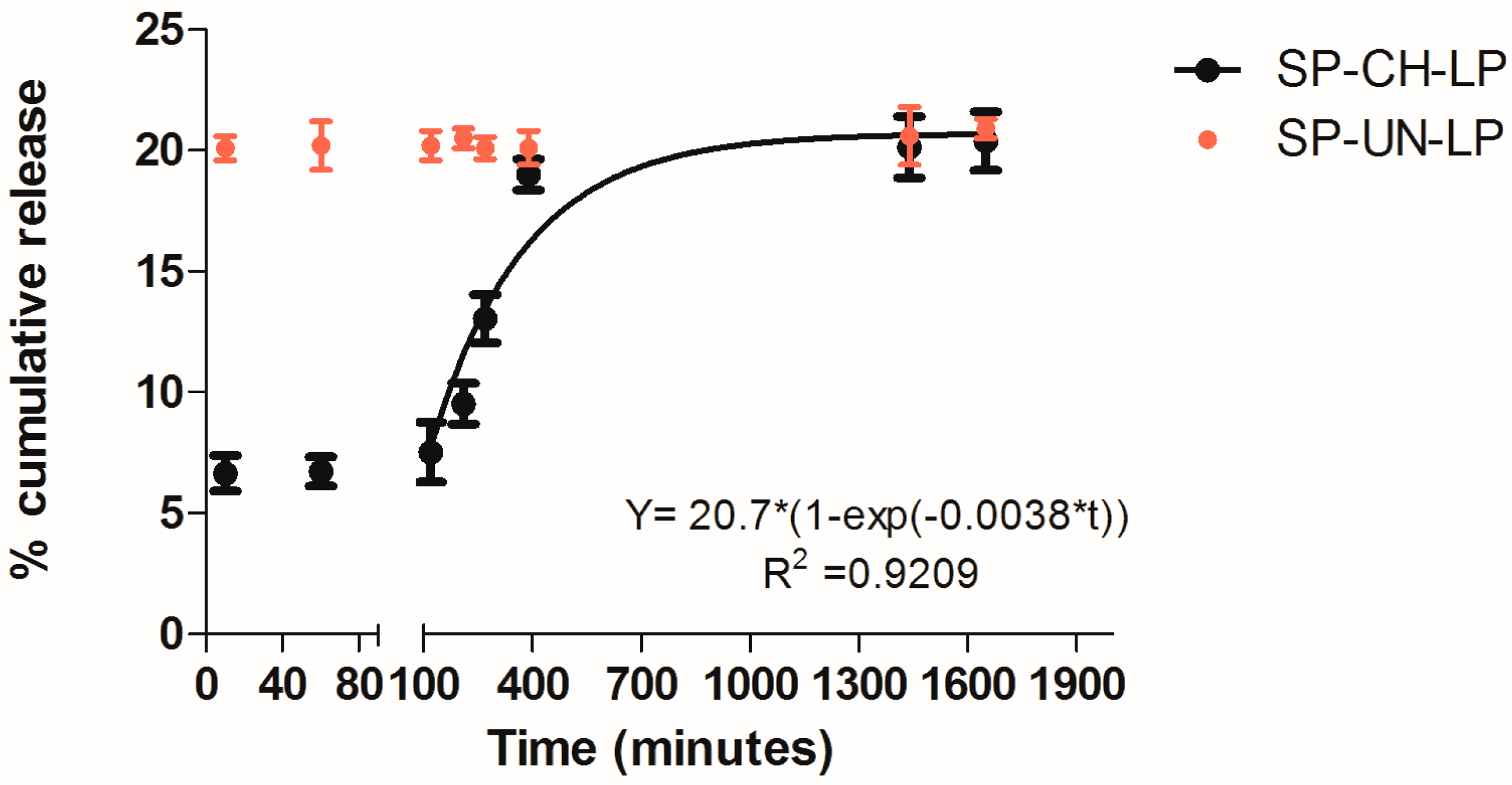
3.2. In Vitro Release Profile
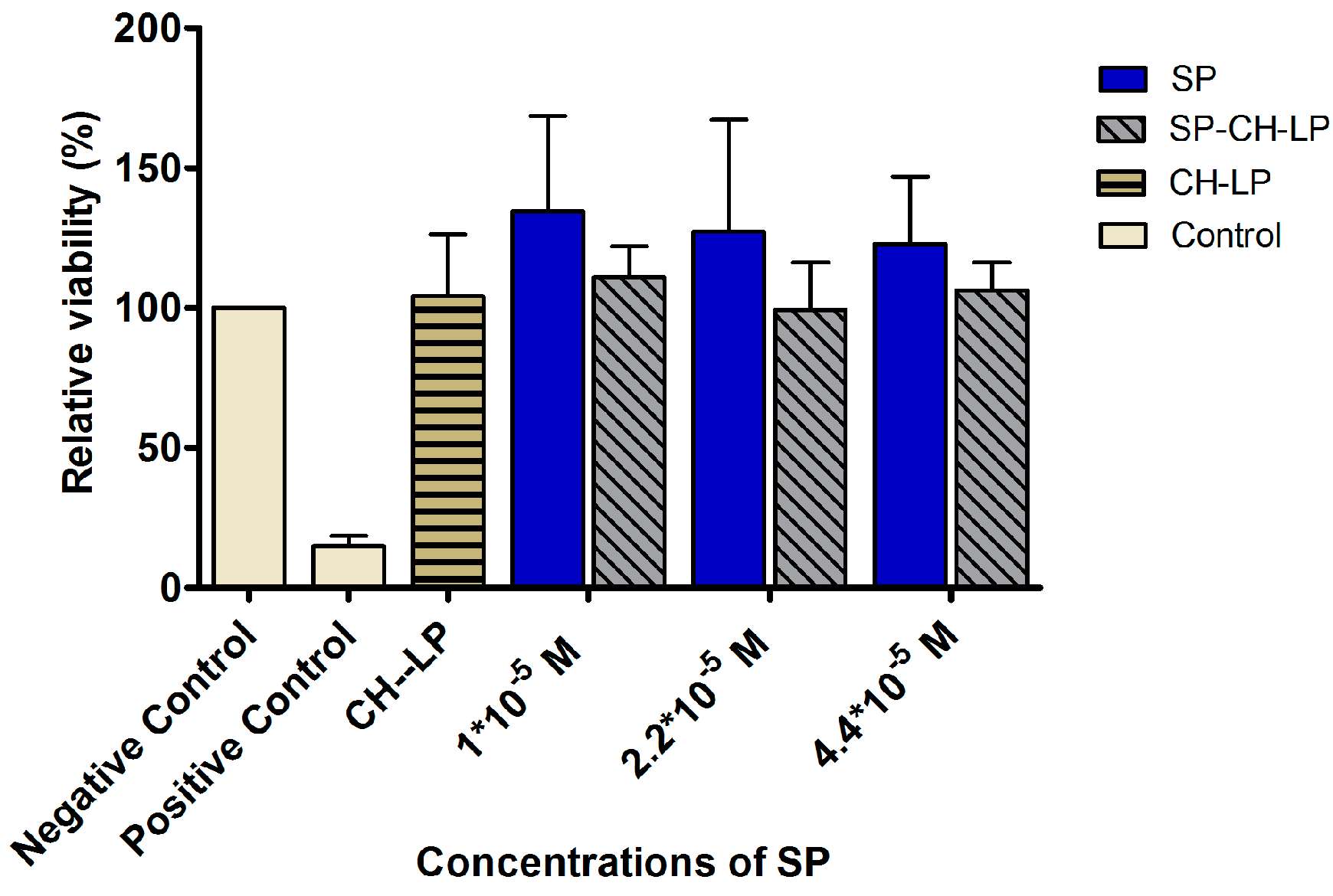
3.3. In Vitro Cytotoxicity
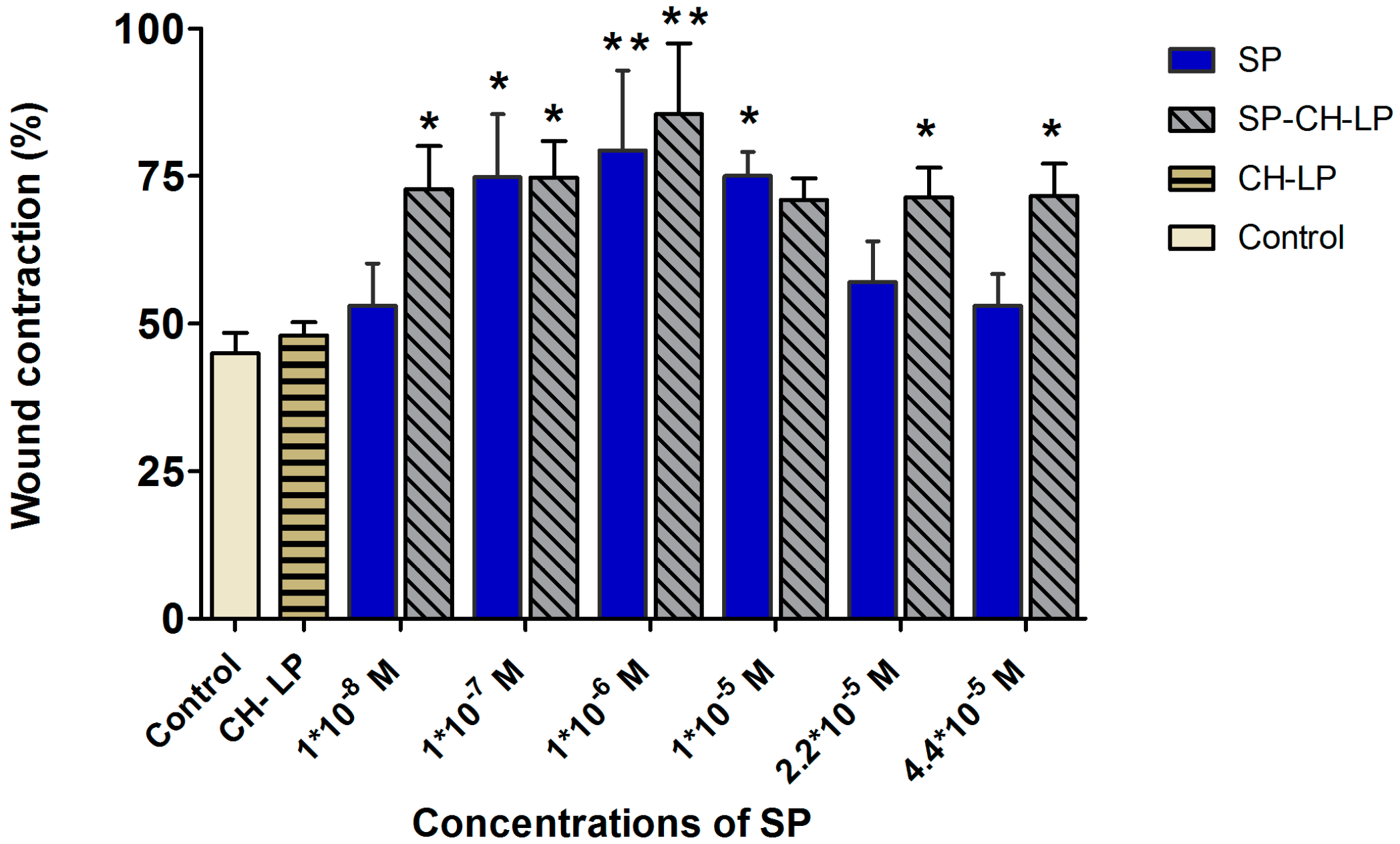
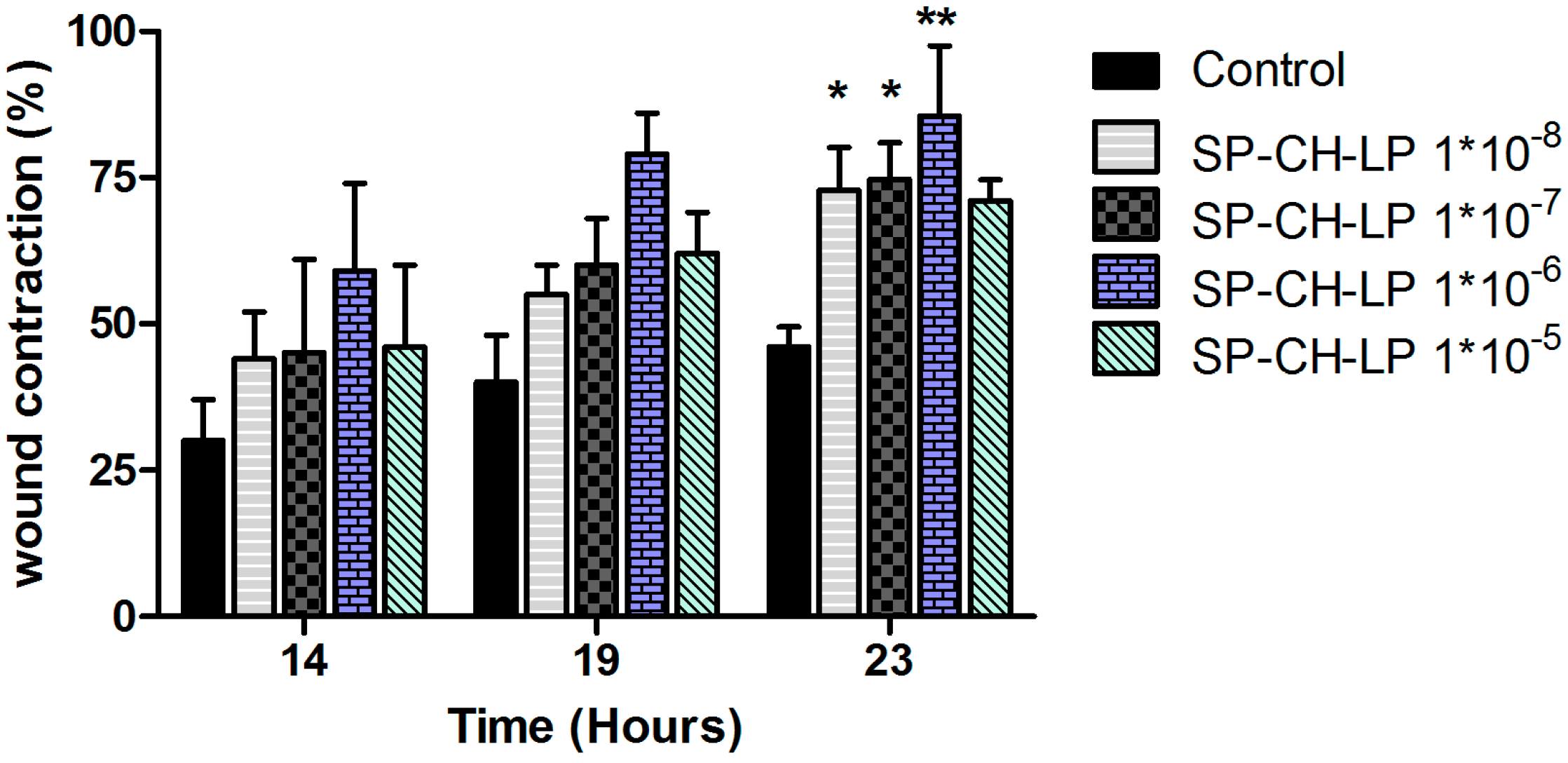
3.4. In Vitro Wound Healing Effect
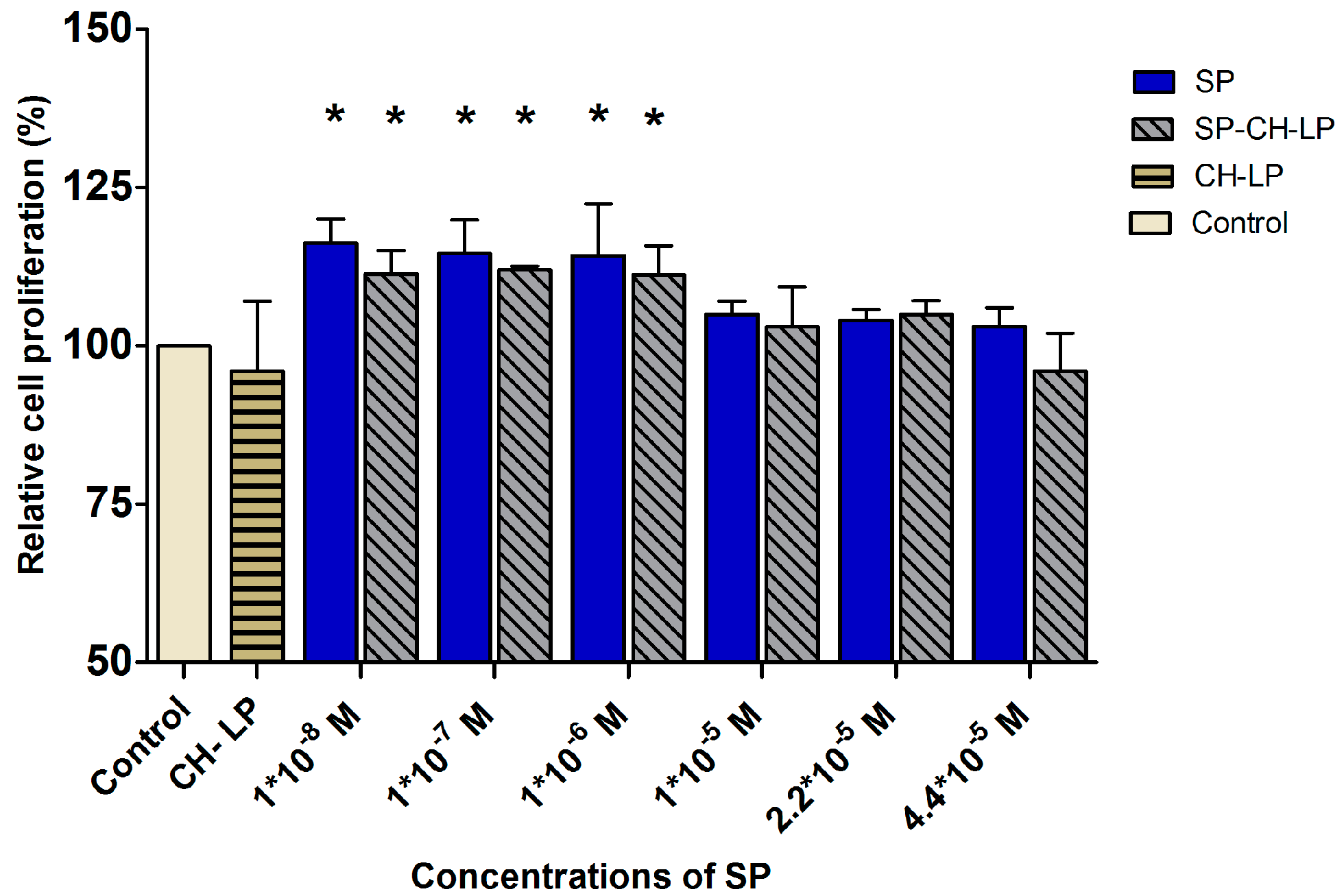
3.5. SP Proliferation Effect on HaCaT Cells
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guo, S.; DiPietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [PubMed]
- Tregear, G.W.; Niall, H.D.; Potts, J.T.; Leeman, S.E.; Chang, M.M. Synthesis of substance P. Nat. New Biol. 1971, 232, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.V.; McManus, A.T.; Chambers, J.P. Exogenous administration of substance P enhances wound healing in a novel skin-injury model. Exp. Biol. Med. (Maywood) 2005, 230, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Kant, V.; Kumar, D.; Kumar, D.; Prasad, R.; Gopal, A.; Pathak, N.N.; Kumar, P.; Tandan, S.K. Topical application of substance P promotes wound healing in streptozotocin-induced diabetic rats. Cytokine 2015, 73, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, T.; Schukraft, J.; Krämer-Best, H.H.; Geber, C.; Ackermann, T.; Birklein, F. Interaction of calcitonin gene related peptide (CGRP) and substance P (SP) in human skin. Neuropeptides 2016, 59, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kohara, H.; Tajima, S.; Yamamoto, M.; Tabata, Y. Angiogenesis induced by controlled release of neuropeptide substance P. Biomaterials 2010, 31, 8617–8625. [Google Scholar] [CrossRef] [PubMed]
- Ziche, M.; Morbidelli, L.; Masini, E.; Amerini, S.; Granger, H.J.; Maggi, C.A.; Geppetti, P.; Ledda, F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J. Clin. Investig. 1994, 94, 2036–2044. [Google Scholar] [CrossRef] [PubMed]
- Muangman, P.; Tamura, R.N.; Muffley, L.A.; Isik, F.F.; Scott, J.R.; Xie, C.; Kegel, G.; Sullivan, S.R.; Liang, Z.; Gibran, N.S. Substance P enhances wound closure in nitric oxide synthase knockout mice. J. Surg. Res. 2009, 153, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, L.; Clark, J.D.; Kingery, W.S. Keratinocytes express cytokines and nerve growth factor in response to neuropeptide activation of the ERK1/2 and JNK MAPK transcription pathways. Regul. Pept. 2013, 186, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Leal, E.C.; Carvalho, E.; Tellechea, A.; Kafanas, A.; Tecilazich, F.; Kearney, C.; Kuchibhotla, S.; Auster, M.E.; Kokkotou, E.; Mooney, D.J.; et al. Substance P promotes wound healing in diabetes by modulating inflammation and macrophage phenotype. Am. J. Pathol. 2015, 185, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell. Mol. Life Sci. 2016, 73, 4249–4264. [Google Scholar] [CrossRef] [PubMed]
- Słoniecka, M.; Le Roux, S.; Zhou, Q.; Danielson, P. Substance P enhances keratocyte migration and neutrophil recruitment through interleukin-8. Mol. Pharmacol. 2016, 89, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Lee, J.; Lee, E.; Kwon, Y.S.; Lee, E.; Ahn, W.; Jiang, M.H.; Kim, J.C.; Son, Y. A new role of substance P as an injury-inducible messenger for mobilization of CD29+ stromal-like cells. Nat. Med. 2009, 15, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Kim, D.Y.; Yoon, K.J.; Son, Y. A new paradigm for stem cell therapy: Substance-P as a stem cell-stimulating agent. Arch. Pharm. Res. 2011, 34, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Burbach, G.J.; Kim, K.H.; Zivony, A.S.; Kim, A.; Aranda, J.; Wright, S.; Naik, S.M.; Caughman, S.W.; Ansel, J.C.; Armstrong, C.A. The neurosensory tachykinins substance P and neurokinin A directly induce keratinocyte nerve growth factor. J. Investig. Dermatol. 2001, 117, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Hochman, B.; Tucci-Viegas, V.M.; Monteiro, P.K.; França, J.P.; Gaiba, S.; Ferreira, L.M. The action of CGRP and SP on cultured skin fibroblasts. Cent. Eur. J. Biol. 2014, 9, 717–726. [Google Scholar] [CrossRef]
- Backman, L.J.; Fong, G.; Andersson, G.; Scott, A.; Danielson, P. Substance P is a mechanoresponsive, autocrine regulator of human tenocyte proliferation. PLoS ONE 2011, 6, e27209. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.; Carvalho, E.; Cruz, M.T. Role of neuropeptides in skin inflammation and its involvement in diabetic wound healing. Expert Opin. Biol. Ther. 2010, 10, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Razavi, R.; Chan, Y.; Afifiyan, F.N.; Liu, X.J.; Wan, X.; Yantha, J.; Tsui, H.; Tang, L.; Tsai, S.; Santamaria, P.; et al. TRPV1+ sensory neurons control β cell stress and islet inflammation in autoimmune diabetes. Cell 2006, 127, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- De Muckadell, O.B.S.; Aggestrup, S.; Stentoft, P. Flushing and plasma substance P concentration during infusion of synthetic substance P in normal man. Scand. J. Gastroenterol. 1986, 21, 498–502. [Google Scholar] [CrossRef]
- Saidi, M.; Kamali, S.; Beaudry, F. Characterization of substance P processing in mouse spinal cord S9 fractions using high-resolution Quadrupole-Orbitrap mass spectrometry. Neuropeptides 2016, 59, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Weglicki, W.B.; Chmielinska, J.J.; Tejero-Taldo, I.; Kramer, J.H.; Spurney, C.F.; Viswalingham, K.; Lu, B.; Tong Mak, I. Neutral endopeptidase inhibition enhances substance P mediated inflammation due to hypomagnesemia. Magnes. Res. 2009, 22, 167–173. [Google Scholar] [CrossRef]
- Probert, L.; Hanley, M.R. The immunocytochemical localisation of “substance-P-degrading enzyme” within the rat spinal cord. Neurosci. Lett. 1987, 78, 132–137. [Google Scholar] [CrossRef]
- Scholzen, T.E.; Luger, T.A. Neutral endopeptidase and angiotensin-converting enzyme—Key enzymes terminating the action of neuroendocrine mediators. Exp. Dermatol. 2004, 13 (Suppl. 4), 22–26. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Izumi, H.; Fukushige, R.; Shimizu, T.; Watanabe, K.; Morioka, N.; Hama, A.; Takamatsu, H.; Nakata, Y. Continuous infusion of substance P into rat striatum alleviates nociceptive behavior via phosphorylation of extracellular signal-regulated kinase 1/2. J. Neurochem. 2015, 131, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Eloy, J.O.; de Souza, M.C.; Petrilli, R.; Barcellos, J.P.A.; Lee, R.J.; Marchetti, J.M. Liposomes as carriers of hydrophilic small molecule drugs: Strategies to enhance encapsulation and delivery. Colloids Surf. B Biointerfaces 2014, 123, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Maitani, Y.; Igarashi, S.; Sato, M.; Hattori, Y. Cationic liposome (DC-Chol/DOPE = 1:2) and a modified ethanol injection method to prepare liposomes, increased gene expression. Int. J. Pharm. 2007, 342, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Gibis, M.; Ruedt, C.; Weiss, J. In vitro release of grape-seed polyphenols encapsulated from uncoated and chitosan-coated liposomes. Food Res. Int. 2016, 88, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Filipović-Grcić, J.; Skalko-Basnet, N.; Jalsenjak, I. Mucoadhesive chitosan-coated liposomes: Characteristics and stability. J. Microencapsul. 2007, 18, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Feng, B.; Zhang, X.; Xia, W.; Xia, S. Biopolymer-coated liposomes by electrostatic adsorption of chitosan (chitosomes) as novel delivery systems for carotenoids. Food Hydrocoll. 2016, 52, 774–784. [Google Scholar] [CrossRef]
- Guo, J. Chitosan-coated liposomes: Characterization and interaction with leuprolide. Int. J. Pharm. 2003, 260, 167–173. [Google Scholar] [CrossRef]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Sorlier, P.; Denuzière, A.; Viton, C.; Domard, A. Relation between the degree of acetylation and the electrostatic properties of chitin and chitosan. Biomacromolecules 2001, 2, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, R.A. Human enzymatic activities related to the therapeutic administration of chitin derivatives. Cell. Mol. Life Sci. 1997, 53, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Menchicchi, B.; Fuenzalida, J.P.; Bobbili, K.B.; Hensel, A.; Swamy, M.J.; Goycoolea, F.M. Structure of chitosan determines its interactions with mucin. Biomacromolecules 2014, 15, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Menchicchi, B.; Fuenzalida, J.P.; Hensel, A.; Swamy, M.J.; David, L.; Rochas, C.; Goycoolea, F.M. Biophysical analysis of the molecular interactions between polysaccharides and mucin. Biomacromolecules 2015, 16, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Mao, H.Q.; Huang, S.K.; Leong, K.W. Oral gene delivery with chitosan—DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat. Med. 1999, 5, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Chen, X.G.; Xing, K.; Park, H.J. Antimicrobial properties of chitosan and mode of action: A state of the art review. Int. J. Food Microbiol. 2010, 144, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Raafat, D.; Von Bargen, K.; Haas, A.; Sahl, H.G. Insights into the mode of action of chitosan as an antibacterial compound. Appl. Environ. Microbiol. 2008, 74, 3764–3773. [Google Scholar] [CrossRef] [PubMed]
- Tokura, S.; Ueno, K.; Miyazaki, S.; Nishi, N. Molecular weight dependent antimicrobial activity by chitosan. New Macromol. Archit. Funct. 1997, 199–207. [Google Scholar] [CrossRef]
- Andersen, T.; Vanić, Ž.; Flaten, G.E.; Mattsson, S.; Tho, I.; Škalko-Basnet, N. Pectosomes and chitosomes as delivery systems for metronidazole: The one-pot preparation method. Pharmaceutics 2013, 5, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Jøraholmen, M.W.; Škalko-Basnet, N.; Acharya, G.; Basnet, P. Resveratrol-loaded liposomes for topical treatment of the vaginal inflammation and infections. Eur. J. Pharm. Sci. 2015, 79, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Cowsik, S.M.; Lücke, C.; Rüterjans, H. Lipid-induced conformation of substance P. J. Biomol. Struct. Dyn. 1997, 15, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Mertins, O.; Dimova, R. Binding of chitosan to phospholipid vesicles studied with isothermal titration calorimetry. Langmuir 2011, 27, 5506–5515. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Yoo, C.Y.; Park, S.N. Improved stability and skin permeability of sodium hyaluronate-chitosan multilayered liposomes by Layer-by-Layer electrostatic deposition for quercetin delivery. Colloids Surf. B Biointerfaces 2015, 129, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.N.; Caride, V.J.; Caldecourt, M.A.; Twickler, J.; Abdullah, V. The mechanism of liposome accumulation in infarction. Biochim. Biophys. Acta (BBA) Gen. Subj. 1984, 797, 363–368. [Google Scholar] [CrossRef]
- Nevalainen, T.J.; Haapamäki, M.M.; Grönroos, J.M. Roles of secretory phospholipases A2 in inflammatory diseases and trauma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2000, 1488, 83–90. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Hu, J.-H.; Zhu, Q.-G.; Li, F.-Q.; Sun, H.-J. Substance P receptor expression in human skin keratinocytes and fibroblasts. Br. J. Dermatol. 2006, 155, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.J.; Lone, A.M.; Tinoco, A.D.; Saghatelian, A. Proteolysis controls endogenous substance P levels. PLoS ONE 2013, 8, e68638. [Google Scholar] [CrossRef] [PubMed]
- Geracioti, T.D.; Carpenter, L.L.; Owens, M.J.; Baker, D.G.; Ekhator, N.N.; Horn, P.S.; Strawn, J.R.; Sanacora, G.; Kinkead, B.; Price, L.H.; et al. Elevated cerebrospinal fluid substance P concentrations in posttraumatic stress disorder and major depression. Am. J. Psychiatry 2006, 163, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Z.; Jin, R.R.; Yang, W.; Xiang, Q.; Yu, W.Z.; Lin, Q.; Tian, F.R.; Mao, K.L.; Lv, C.Z.; Wáng, Y.X.J.; et al. Using gelatin nanoparticle mediated intranasal delivery of neuropeptide substance P to enhance neuro-recovery in hemiparkinsonian rats. PLoS ONE 2016, 11, e0148848. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.J.; Tublitz, N.J.; Minson, C.T. Neurokinin-1 receptor desensitization to consecutive microdialysis infusions of substance P in human skin. J. Physiol. 2005, 568, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Sugiya, H.; Tennes, K.A.; Putney, J.W., Jr. Homologous desensitization of substance-P-induced inositol polyphosphate formation in rat parotid acinar cells. Biochem. J. 1987, 244, 647–653. [Google Scholar] [CrossRef] [PubMed]
- McConalogue, K.; Corvera, C.U.; Gamp, P.D.; Grady, E.F.; Bunnett, N.W. Desensitization of the neurokinin-1 receptor (NK1-R) in neurons: Effects of substance P on the distribution of NK1-R, Galphaq/11, G-protein receptor kinase-2/3 and beta-arrestin-1/2. Mol. Biol. Cell 1998, 9, 2305–2324. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.N.; Goldstein, B.D.; Aronstam, R.S. Substance P receptor desensitization in the dorsal horn: possible involvement of receptor-G protein complexes. Brain Res. 1993, 600, 89–96. [Google Scholar] [CrossRef]
- Kim, K.-T.; Kim, H.-J.; Cho, D.-C.; Bae, J.-S.; Park, S.-W. Substance P stimulates proliferation of spinal neural stem cells in spinal cord injury via the mitogen-activated protein kinase signaling pathway. Spine J. 2015, 15, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Roggenkamp, D.; Köpnick, S.; Stäb, F.; Wenck, H.; Schmelz, M.; Neufang, G. Epidermal nerve fibers modulate keratinocyte growth via neuropeptide signaling in an innervated skin model. J. Investig. Dermatol. 2013, 133, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Kant, V.; Kumar, D.; Prasad, R.; Gopal, A.; Pathak, N.N.; Kumar, P.; Tandan, S.K. Combined effect of substance P and curcumin on cutaneous wound healing in diabetic rats. J. Surg. Res. 2017, 212, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Um, J.; Yu, J.; Park, K. Substance P accelerates wound healing in type 2 diabetic mice through endothelial progenitor cell mobilization and Yes-associated protein activation. Mol. Med. Rep. 2017, 3035–3040. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Size (d.,nm) 1 | PdI 1 | ζ (mV) 2 | EE (%) 3 |
|---|---|---|---|---|
| SP-UN-LP | 151 ± 27 | 0.2 ± 0.06 | −49 ± 2.5 | 81.3 ± 6 |
| UN-LP | 104 ± 9 | 0.2 ± 0.01 | −60 ± 1.3 | |
| SP-CH-LP | 243 ± 24 | 0.3 ± 0.1 | +32 ± 1.0 | 66 ± 3.5 |
| CH-LP | 247 ± 26 | 0.2 ± 0.06 | +30 ± 2.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mengoni, T.; Adrian, M.; Pereira, S.; Santos-Carballal, B.; Kaiser, M.; Goycoolea, F.M. A Chitosan—Based Liposome Formulation Enhances the In Vitro Wound Healing Efficacy of Substance P Neuropeptide. Pharmaceutics 2017, 9, 56. https://doi.org/10.3390/pharmaceutics9040056
Mengoni T, Adrian M, Pereira S, Santos-Carballal B, Kaiser M, Goycoolea FM. A Chitosan—Based Liposome Formulation Enhances the In Vitro Wound Healing Efficacy of Substance P Neuropeptide. Pharmaceutics. 2017; 9(4):56. https://doi.org/10.3390/pharmaceutics9040056
Chicago/Turabian StyleMengoni, Tamara, Manuela Adrian, Susana Pereira, Beatriz Santos-Carballal, Mathias Kaiser, and Francisco M. Goycoolea. 2017. "A Chitosan—Based Liposome Formulation Enhances the In Vitro Wound Healing Efficacy of Substance P Neuropeptide" Pharmaceutics 9, no. 4: 56. https://doi.org/10.3390/pharmaceutics9040056
APA StyleMengoni, T., Adrian, M., Pereira, S., Santos-Carballal, B., Kaiser, M., & Goycoolea, F. M. (2017). A Chitosan—Based Liposome Formulation Enhances the In Vitro Wound Healing Efficacy of Substance P Neuropeptide. Pharmaceutics, 9(4), 56. https://doi.org/10.3390/pharmaceutics9040056