
Alignment of Short Reads: A Crucial Step for Application of Next-Generation Sequencing Data in Precision Medicine



Abstract
:
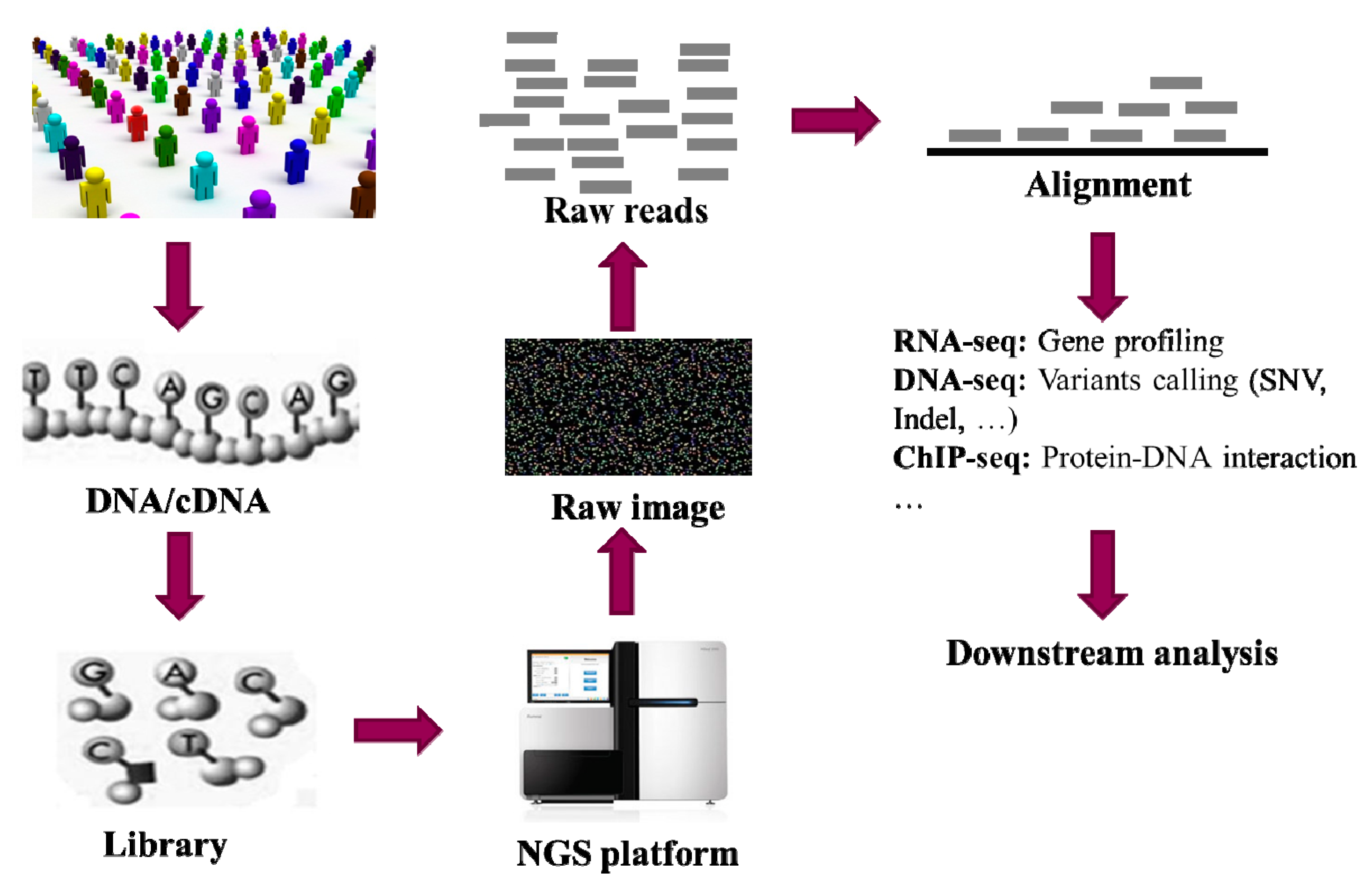
1. Introduction
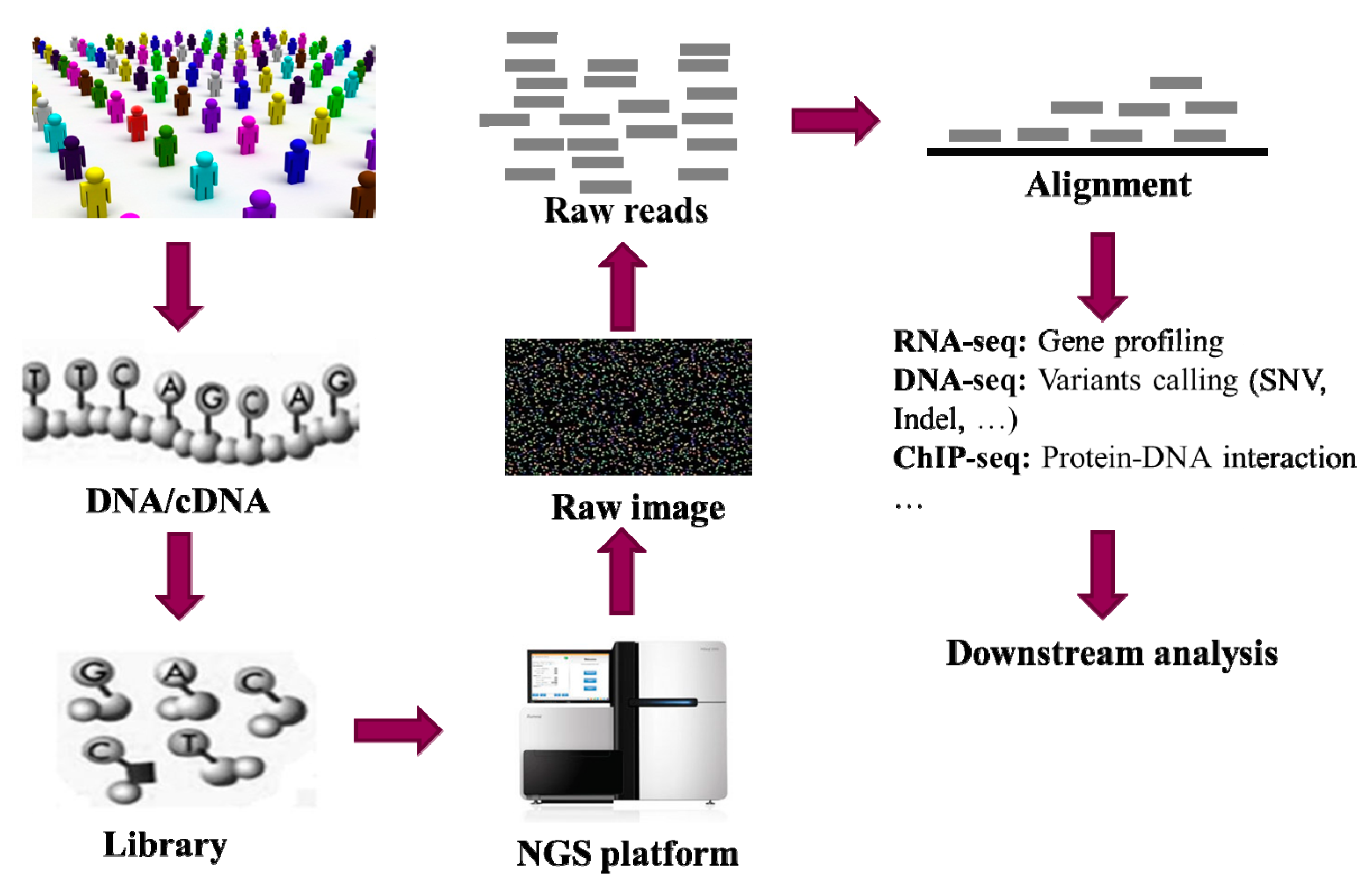

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform Name | Illumina HiSeq 2500 | Ion Torrent-Proton II | PacBio RS II | OxFord Nanopore Minion |
|---|---|---|---|---|
| Instrument |  |  |  |  |
| Cost (USD) ** | 690 k | 224 k | 695 k | 1 k *** |
| Reagent cost Per run/per GB | 4126/45.84 | 1000/20.41 | 100/1111.11 | 900/1000 |
| Reads per run | 300 millions | 280 millions | 0.03 millions | 0.1 millions |
| Average Read length | 2 × 150 bp | 175 bp | 14,000 bp | 9,000 bp |
| Run time | 10 h | 5 h | 2 h | 6 h |
| Major errors | substitution | indel | indel | deletion |
| Error rate (%) | 0.1 | 1 | 1 | 4 |
| Amplification | bridgePCR | emPCR | none, SMS | none, SMS |
| Advantage | low cost per GB; high output | low cost | long reads; no amplification bias | long reads; no amplification bias |
| Disadvantage | high cost | homopolymer errors | low throughput; high cost | high error rate |
| Name | Website | Reference | Remark |
|---|---|---|---|
| SOAP * | soap.genomics.org.cn | [32,33,34,35] | k-mer inexact match seed; support at most 3 mismatches; GPU calculation supported |
| CUSHAW $ | cushaw3.sourceforge.net/homepage.htm#downloads | [36,37,38,39] | k-mer inexact match, maximal exact match and hybrid seeds; GPU supported |
| Bowtie & | bowtie-bio.sourceforge.net | [40,41] | k-mer inexact match seed; high speed; double-index; up to 3 mismatches |
| BWA | bio-bwa.sourceforge.net | [42,43] | k-mer inexact match and maximal exact match seed |
| GASSST | www.irisa.fr/symbiose/projects/gassst/ | [44] | k-mer exact match seed; it currently has been tested for reads up to 500 bp |
| GNUMAP | dna.cs.byu.edu/gnumap/ | [45] | k-mer exact match seed; probabilistically mapping reads to repeat regions |
| MOSAIK | gkno.me/pipelines.html#mosaik | [46] | k-mer exact match seed |
| NextGenMap | cibiv.github.io/NextGenMap/ | [47] | q-gramq-gram filter; GPU calculation supported |
| QPALMA | www.raetschlab.org/suppl/qpalma | [48] | k-mer inexact match; incorporate read quality score and splice site |
| RMAP | rulai.cshl.edu/rmap/ | [49,50] | k-mer inexact match seed; 10 mismatches allowed; incorporate read quality score |
| Segemehl | www.bioinf.uni-leipzig.de/Software/segemehl/ | [51] | k-mer inexact match seed; enhanced suffix arrays |
| SeqMap | www-personal.umich.edu/ ~jianghui/seqmap/ | [52] | k-mer inexact match; support windows, linux, Mac OS |
| Stampy | www.well.ox.ac.uk/project-stampy | [53] | k-mer inexact match; support up to 30 bp indels in paired-end reads alignment |
| Cloudburst | sourceforge.net/projects/cloudburst-bio/ | [54] | Highly sensitive read mapping with MapReduce. |
| drFAST | drfast.sourceforge.net/ | [55] | k-mer inexact match; specially designed for better delineation of structural variants |
| BFAST | sourceforge.net/projects/bfast/ | [56] | k-mer spaced seeds |
| MAQ | maq.sourceforge.net | [57] | k-mer spaced seeds; incorporate quality scores of reads in alignment |
| MOM | go.vcu.edu/mom | [58] | k-mer spaced seeds; unlimited mismatches in the 3′ and 5′ flanking regions. |
| PASS | pass.cribi.unipd.it | [59] | k-mer spaced seeds; implemented in C++ and supported on Linux and Windows |
| PerM | code.google.com/p/perm/ | [60] | k-mer spaced seeds; 9 mismatches are allowed |
| SHRiMP2 | compbio.cs.toronto.edu/shrimp/ | [61,62] | combined k-mer spaced seeds and q-gram filter |
| ZOOM | www.bioinfor.com/zoom/general/overview.html | [63] | k-mer spaced seeds; tolerate 2 mismatches by default |
| BarraCUDA | seqbarracuda.sourceforge.net/ | [64] | Incorporate GPU to speed up BWA |
| GEM | gemlibrary.sourceforge.net/ | [65] | q-gram filter |
| MPSCAN | www.atgc-montpellier.fr/mpscan/ | [66] | q-gram filter; support Windows, linux, Mac OS |
| ERNE | iga-rna.sourceforge.net/ | [67] | long gap support; Works on Windows, Mac OS X, linux |
| SARUMAN | www.cebitec.uni-bielefeld.de/ brf/saruman/saruman.html | [68] | k-mer inexact matched seed; support GPU calculation |
| LAST | last.cbrc.jp/ | [69] | adaptive seed |
| Genalice MAP | www.genalice.com/product/genalice-map/ | NA | cloud calculation; High sensitivity for SNPs and long INDELS |
| Novoalign | www.novocraft.com/ | NA | support up to 7 and 16 mismatches in single-end and pair-end reads. |
| PRIMEX | bioinformatics.cribi.unipd.it/primex | [70] | k-mer inexact match seed; written in C++; lookup table and server functionality |
| SOCS | solidsoftwaretools.com/gf/project/socs/ | [71] | good at align CpG methylation-enriched reads |
| SToRM | bioinfo.lifl.fr/yass/iedera_solid/storm/ | [72] | doesn’t support pair-end reads |
| iSAAC | https://github.com/sequencing/isaac_aligner | [73] | k-mer inexact match seed; high speed |
| RazerS | www.seqan.de/projects/razers/ | [74] | q-gram filter; support Windows, linux, Mac OS X |
| SSAHA2 | www.sanger.ac.uk/resources/software/ssaha2/ | [75] | k-mer inexact match seed; support various output formats |
| UGENE | ugene.unipro.ru/ | [76] | works on Windows, linux and Mac OS X |
2. Strategies of Current Alignment Algorithms
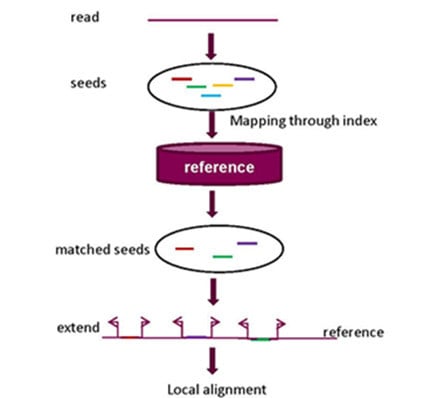
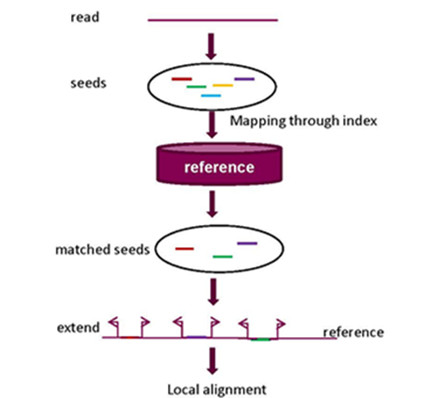
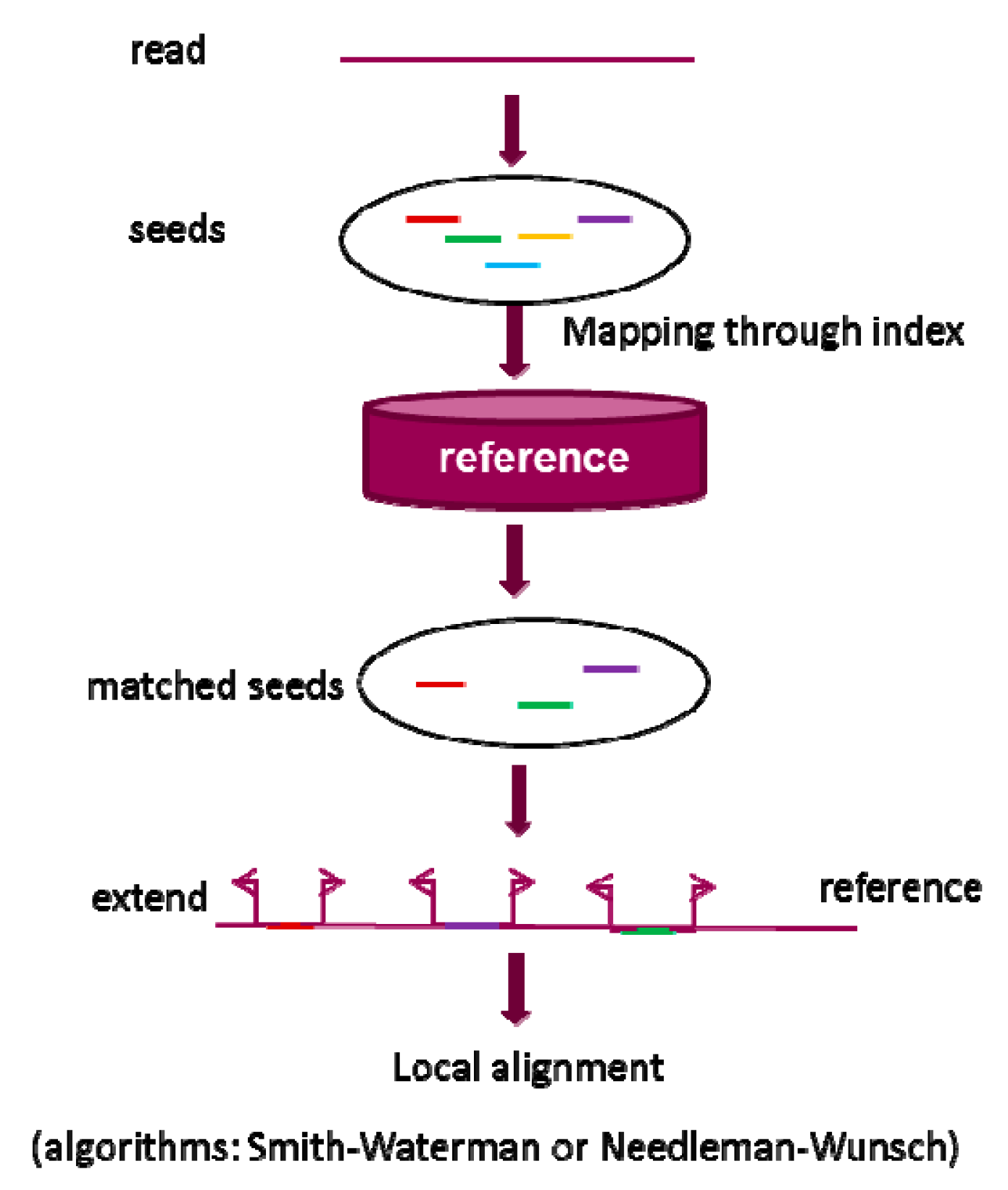
2.1. Seed-and-Extend Strategy
2.1.1. k-mer Exact Match Seed
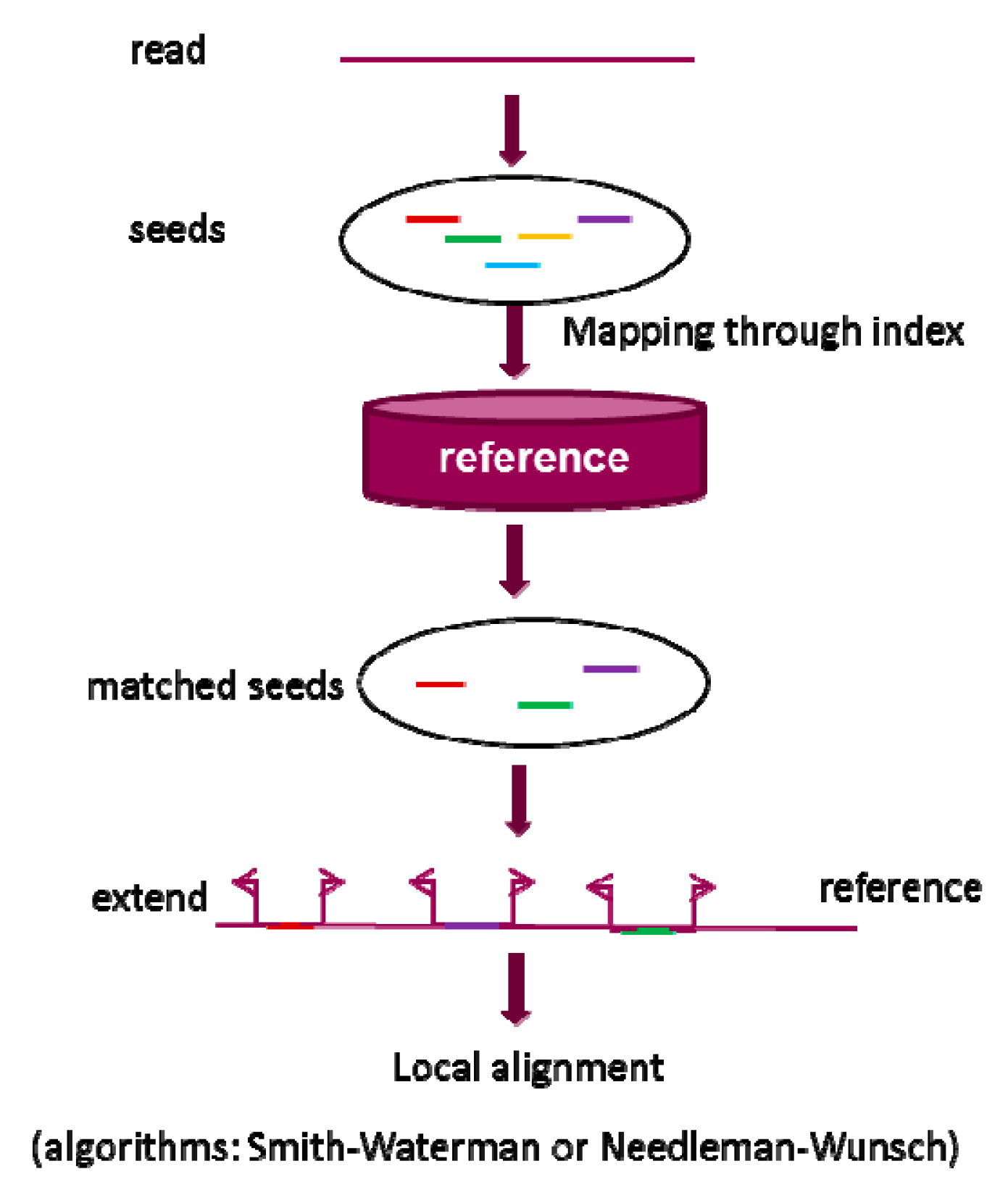
2.1.2. k-mer Inexact Match Seed
2.1.3. k-mer Spaced Seed
2.1.4. Maximum Extend Match (MEM) Seed
2.1.5. Adaptive Seed
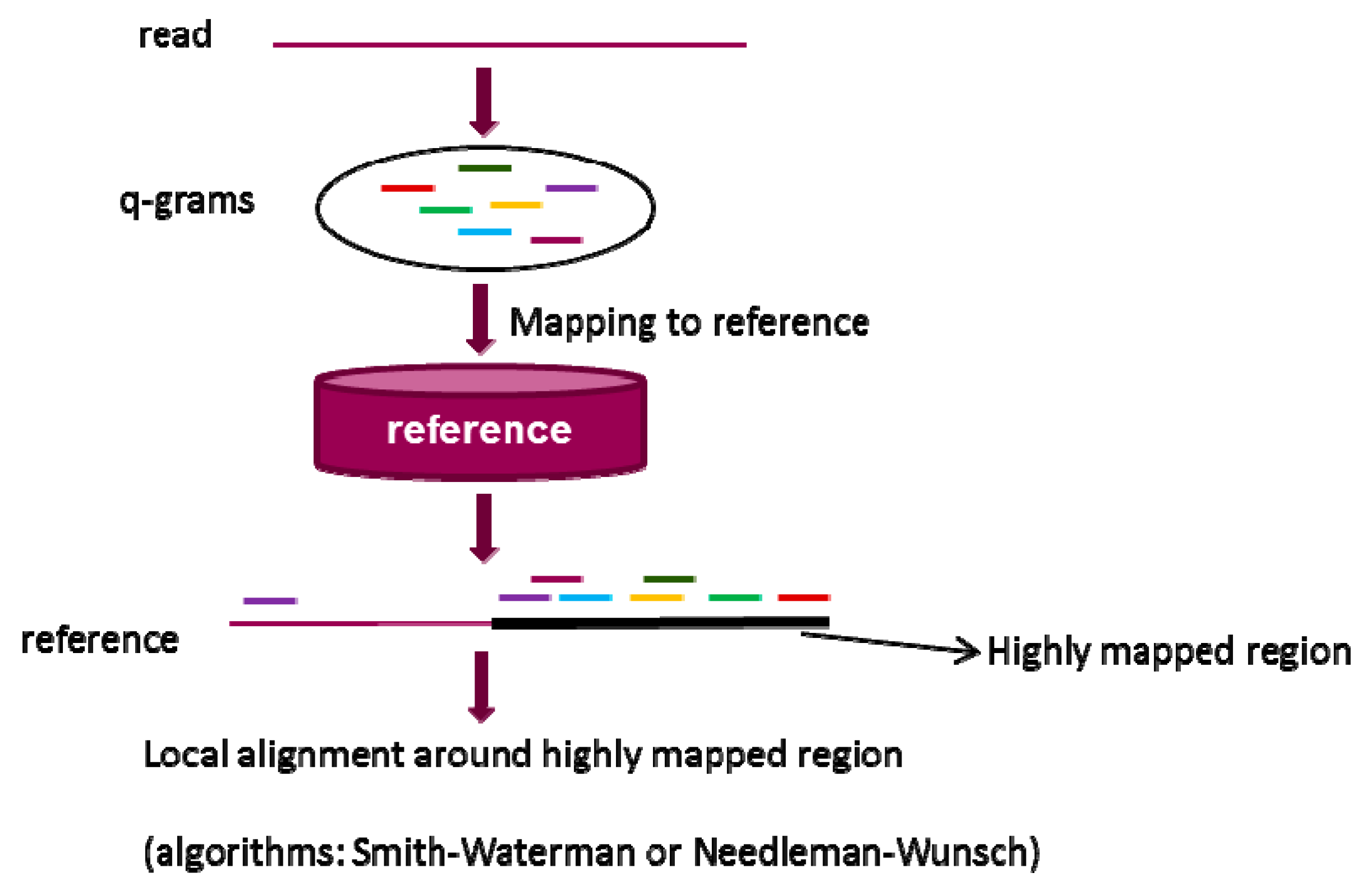
2.2. q-gram Filter
3. Conclusions
4. Further Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Eng. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.J. The success of precision medicine requires a public health perspective. Available online: http://blogs.cdc.gov/genomics/2015/01/29/precision-medicine/ (accessed on 17 November 2015).
- Hong, H.; Goodsaid, F.; Shi, L.; Tong, W. Molecular biomarkers: A US FDA effort. Biomark. Med. 2010, 4, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Slikker, W., Jr. Advancing translation of biomarkers into regulatory decision making. Biomark. Med. 2015, 9, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Hong, H.; Perkins, E.J. Ionotropic GABA Receptor Antagonism-Induced Adverse Outcome Pathways for Potential Neurotoxicity Biomarkers. Biomark. Med. 2015, 9, 1225–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hong, H.; Mendrick, D.L.; Tang, T.; Cheng, F. Biomarker-based Drug Safety Assessment in the Age of Systems Pharmacology: From Foundational to Regulatory Science. Biomark. Med. 2015, 9, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.; Zou, W.; Hong, H.; Fang, H.; Tong, W. Molecular Regulation of miRNAs and Potential Biomarkers in the Progression of Hepatic Steatosis. Biomark. Med. 2015, 9, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Tolleson, W.H.; Guo, L.; Yu, D.; Chen, S.; Hong, H.; Mattes, W.; Ning, B. MicroRNAs as Pharmacogenomic Biomarkers for Drug Efficacy and Drug Safety Assessment. Biomark. Med. 2015, 9, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tong, W. Emerging efforts for discovering new biomarkers of liver disease and hepatotoxicity. Biomark. Med. 2014, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Xu, L.; Liu, J.; Jones, W.D.; Su, Z.; Ning, B.; Perkins, R.; Ge, W.; Miclaus, K.; Zhang, L.; et al. Technical reproducibility of genotyping snp arrays used in genome-wide association studies. PLoS ONE 2012, 7, e44483. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Shi, L.; Su, Z.; Ge, W.; Jones, W.; Czika, W.; Miclaus, K.; Lambert, C.; Vega, S.; Zhang, J.; et al. Assessing sources of inconsistencies in genotypes and their effects on genome-wide association studies with hapmap samples. Pharmacogenomics J. 2010, 10, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Su, Z.; Ge, W.; Shi, L.; Perkins, R.; Fang, H.; Xu, J.; Chen, J.J.; Han, T.; Kaput, J.; et al. Assessing batch effects of genotype calling algorithm brlmm for the affymetrix genechip human mapping 500 k array set using 270 hapmap samples. BMC Bioinforma. 2008, 9, S17. [Google Scholar] [CrossRef] [PubMed]
- Miclaus, K.; Wolfinger, R.; Vega, S.; Chierici, M.; Furlanello, C.; Lambert, C.; Hong, H.; Zhang, L.; Yin, S.; Goodsaid, F. Batch effects in the brlmm genotype calling algorithm influence gwas results for the affymetrix 500k array. Pharmacogenomics J. 2010, 10, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Schumacher, M.; Scherer, A.; Sanoudou, D.; Megherbi, D.; Davison, T.; Shi, T.; Tong, W.; Shi, L.; Hong, H.; et al. A comparison of batch effect removal methods for enhancement of cross-batch prediction performance using MAQC-II microarray gene expression data. Pharmacogenomics J. 2010, 10, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Shi, L.; Fuscoe, J.C.; Goodsaid, F.; Mendrick, D.; Tong, W. Potential sources of spurious associations and batch effects in genome-wide association studies. In Batch Effects and Noise in Microarray Experiments: Sources and Solutions; Scherer, A., Ed.; John Wiley & Sons: West Sussex, UK, 2009; pp. 191–201. [Google Scholar]
- Hong, H.; Su, Z.; Ge, W.; Shi, L.; Perkins, R.; Fang, H.; Mendrick, D.; Tong, W. Evaluating variations of genotype calling: A potential source of spurious associations in genome-wide association studies. J. Genetics 2010, 89, 55–64. [Google Scholar] [CrossRef]
- Zhang, L.; Yin, S.; Miclaus, K.; Chierici, M.; Vega, S.; Lambert, C.; Hong, H.; Wolfinger, R.; Furlanello, C.; Goodsaid, F. Assessment of Variability in GWAS with CRLMM Genotyping Algorithm on WTCCC Coronary Artery Disease. Pharmacogenomics J. 2010, 10, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Miclaus, K.; Chierici, M.; Lambert, C.; Zhang, L.; Vega, S.; Hong, H.; Yin, S.; Furlanello, C.; Wolfinger, R.; Goodsaid, F. Variability in GWAS Analysis: the Impact of Genotype Calling Algorithm Inconsistencies. Pharmacogenomics J. 2010, 10, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Meehan, J.; Su, Z.; Ng, H.W.; Shu, M.; Luo, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Whole genome sequencing of 35 individuals provides insights into the genetic architecture of korean population. BMC Bioinforma. 2014, 15, S6. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jennings, S.F.; Tong, W.; Hong, H. Next generation sequencing for profiling expression of miRNAs: Technical progress and applications in drug development. J. Biomed. Sci. Eng. 2011, 4, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Ning, B.; Fang, H.; Hong, H.; Perkins, R.; Tong, W.; Shi, L. Next-generation sequencing and its applications in molecular diagnostics. Expert Rev. Mol. Diagn. 2011, 11, 333–343. [Google Scholar] [PubMed]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Soika, V.; Meehan, J.; Su, Z.; Ge, W.; Ng, H.; Perkins, R.; Simonyan, V.; Tong, W.; Hong, H. Quality control metrics improve repeatability and reproducibility of single-nucleotide variants derived from whole-genome sequencing. Pharmacogenomics J. 2015, 15, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Dragan, Y.; Epstein, J.; Teitel, C.; Chen, B.; Xie, Q.; Fang, H.; Shi, L.; Perkins, R.; Tong, W. Quality control and quality assessment of data from surface-enhanced laser desorption/ionization (SELDI) time-of flight (TOF) mass spectrometry (MS). BMC Bioinforma. 2005, 6, S5. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Qiu, J.; Hayden, E.C. Genomics sizes up. Nature 2008, 451, 234. [Google Scholar] [CrossRef] [PubMed]
- Regalado, A. Emtech: Illumina says 228,000 human genomes will be sequenced this year. Available online: http://www.technologyreview.com/news/531091/emtech-illumina-says-228000-human-genomes-will-be-sequenced-this-year/ (accessed on 17 November 2015).
- Bioethics news. Available online: http://www.bioethics.net/news/emtech-illumina-says-228000-human-genomes-will-be-sequenced-this-year/ (accessed on 20 November 2015).
- Hong, H.; Zhang, W.; Shen, J.; Su, Z.; Ning, B.; Han, T.; Perkins, R.; Shi, L.; Tong, W. Critical role of bioinformatics in translating huge amounts of next-generation sequencing data into personalized medicine. Sci. China Life Sci. 2013, 56, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Su, Z.; Mei, N.; Hong, H.; Deng, H.; Shi, L.; Fuscoe, J.C.; Tolleson, W.H. Toxicogenomics and cancer susceptibility: advances with next-generation sequencing. J. Environ. Sci. Health Part C 2014, 32, 121–158. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Salzberg, S.L. How to map billions of short reads onto genomes. Nature Biotechnol. 2009, 27, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. Soap: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, J. Soap2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-M.; Wong, T.; Wu, E.; Luo, R.; Yiu, S.-M.; Li, Y.; Wang, B.; Yu, C.; Chu, X.; Zhao, K. Soap3: Ultra-fast gpu-based parallel alignment tool for short reads. Bioinformatics 2012, 28, 878–879. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Wong, T.; Zhu, J.; Liu, C.-M.; Zhu, X.; Wu, E.; Lee, L.-K.; Lin, H.; Zhu, W.; Cheung, D.W.; et al. Soap3-dp: Fast, accurate and sensitive gpu-based short read aligner. PLoS ONE 2013, 8, e65632. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schmidt, B.; Maskell, D.L. Cushaw: A cuda compatible short read aligner to large genomes based on the burrows-wheeler transform. Bioinformatics 2012, 28, 1830–1837. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schmidt, B. Long read alignment based on maximal exact match seeds. Bioinformatics 2012, 28, i318–i324. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schmidt, B. Cushaw2-GPU: Empowering faster gapped short-read alignment using GPU computing. Design Test IEEE 2014, 31, 31–39. [Google Scholar]
- Liu, Y.; Popp, B.; Schmidt, B. Cushaw3: Sensitive and accurate base-space and color-space short-read alignment with hybrid seeding. PLoS ONE 2014, 9, e86869. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nature Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Li, D.G.; Lin, H.; Li, L.; Li, X.; Liu, Z.C. The correlation of gene expression and co-regulated gene patterns in characteristic kegg pathways. J. Theor. Biol. 2010, 266, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Rizk, G.; Lavenier, D. Gassst: Global alignment short sequence search tool. Bioinformatics 2010, 26, 2534–2540. [Google Scholar] [CrossRef] [PubMed]
- Clement, N.L.; Snell, Q.; Clement, M.J.; Hollenhorst, P.C.; Purwar, J.; Graves, B.J.; Cairns, B.R.; Johnson, W.E. The gnumap algorithm: Unbiased probabilistic mapping of oligonucleotides from next-generation sequencing. Bioinformatics 2010, 26, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-P.; Stromberg, M.P.; Ward, A.; Stewart, C.; Garrison, E.P.; Marth, G.T. Mosaik: A hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS ONE 2014, 9, e90581. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; von Haeseler, A. NextGenMap: Fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 2013, 29, 2790–2791. [Google Scholar] [CrossRef] [PubMed]
- De Bona, F.; Ossowski, S.; Schneeberger, K.; Rätsch, G. Optimal spliced alignments of short sequence reads. Bioinformatics 2008, 24, i174–i180. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Chung, W.-Y.; Hodges, E.; Kendall, J.; Hannon, G.; Hicks, J.; Xuan, Z.; Zhang, M.Q. Updates to the rmap short-read mapping software. Bioinformatics 2009, 25, 2841–2842. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Xuan, Z.; Zhang, M.Q. Using quality scores and longer reads improves accuracy of solexa read mapping. BMC Bioinforma 2008, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermüller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wong, W.H. Seqmap: Mapping massive amount of oligonucleotides to the genome. Bioinformatics 2008, 24, 2395–2396. [Google Scholar] [CrossRef] [PubMed]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.C. Cloudburst: Highly sensitive read mapping with mapreduce. Bioinformatics 2009, 25, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; Hach, F.; Sahinalp, S.C.; Eichler, E.E.; Alkan, C. Sensitive and fast mapping of di-base encoded reads. Bioinformatics 2011, 27, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Homer, N.; Merriman, B.; Nelson, S.F. Bfast: An alignment tool for large scale genome resequencing. PLoS ONE 2009, 4, e7767. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Eaves, H.L.; Gao, Y. Mom: Maximum oligonucleotide mapping. Bioinformatics 2009, 25, 969–970. [Google Scholar] [CrossRef] [PubMed]
- Campagna, D.; Albiero, A.; Bilardi, A.; Caniato, E.; Forcato, C.; Manavski, S.; Vitulo, N.; Valle, G. Pass: A program to align short sequences. Bioinformatics 2009, 25, 967–968. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Souaiaia, T.; Chen, T. Perm: Efficient mapping of short sequencing reads with periodic full sensitive spaced seeds. Bioinformatics 2009, 25, 2514–2521. [Google Scholar] [CrossRef] [PubMed]
- Rumble, S.M.; Lacroute, P.; Dalca, A.V.; Fiume, M.; Sidow, A.; Brudno, M. SHRiMP: Accurate mapping of short color-space reads. PLoS Comput. Biol. 2009, 5, e1000386. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Dzamba, M.; Lister, D.; Ilie, L.; Brudno, M. SHRiMP: Sensitive yet practical short read mapping. Bioinformatics 2011, 27, 1011–1012. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhang, Z.; Zhang, M.; Ma, B.; Li, M. ZOOM! Zillions of oligos mapped. Bioinformatics 2008, 24, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- Klus, P.; Lam, S.; Lyberg, D.; Cheung, M.S.; Pullan, G.; McFarlane, I.; Yeo, G.S.; Lam, B.Y. Barracuda-a fast short read sequence aligner using graphics processing units. BMC Res. Notes 2012, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The gem mapper: Fast, accurate and versatile alignment by filtration. Nature Methods 2012, 9, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Rivals, E.; Salmela, L.; Kiiskinen, P.; Kalsi, P.; Tarhio, J. Mpscan: Fast localisation of multiple reads in genomes. In Algorithms in bioinformatics; Springer: Philadelphia, PA, USA, 2009; pp. 246–260. [Google Scholar]
- Prezza, N.; Del Fabbro, C.; Vezzi, F.; De Paoli, E.; Policriti, A. Erne-bs5: Aligning bs-treated sequences by multiple hits on a 5-letters alphabet. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, New York, NY, USA, 8–10 October 2012; pp. 12–19.
- Blom, J.; Jakobi, T.; Doppmeier, D.; Jaenicke, S.; Kalinowski, J.; Stoye, J.; Goesmann, A. Exact and complete short-read alignment to microbial genomes using graphics processing unit programming. Bioinformatics 2011, 27, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Lexa, M.; Valle, G. Primex: Rapid identification of oligonucleotide matches in whole genomes. Bioinformatics 2003, 19, 2486–2488. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Cochran, C.; Landers, M.; Meredith, G.D.; Dudas, M.; Bergman, N.H. An alignment algorithm for bisulfite sequencing using the applied biosystems solid system. Bioinformatics 2010, 26, 1901–1902. [Google Scholar] [CrossRef] [PubMed]
- Noé, L.; Gîrdea, M.; Kucherov, G. Designing efficient spaced seeds for solid read mapping. Adv. Bioinforma. 2010, 2010, 708501. [Google Scholar] [CrossRef] [PubMed]
- Raczy, C.; Petrovski, R.; Saunders, C.T.; Chorny, I.; Kruglyak, S.; Margulies, E.H.; Chuang, H.-Y.; Källberg, M.; Kumar, S.A.; Liao, A. Isaac: Ultra-fast whole-genome secondary analysis on illumina sequencing platforms. Bioinformatics 2013, 29, 2041–2043. [Google Scholar] [CrossRef] [PubMed]
- Weese, D.; Holtgrewe, M.; Reinert, K. Razers 3: Faster, fully sensitive read mapping. Bioinformatics 2012, 28, 2592–2599. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Cox, A.J.; Mullikin, J.C. Ssaha: A fast search method for large DNA databases. Genome Res. 2001, 11, 1725–1729. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro ugene: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Flicek, P.; Birney, E. Sense from sequence reads: Methods for alignment and assembly. Nature Methods 2009, 6, S6–S12. [Google Scholar] [CrossRef] [PubMed]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Blumer, A.; Blumer, J.; Haussler, D.; Ehrenfeucht, A.; Chen, M.-T.; Seiferas, J. The smallest automation recognizing the subwords of a text. Theor. Computer Sci. 1985, 40, 31–55. [Google Scholar] [CrossRef]
- Li, H.; Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinforma. 2010, 11, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Tromp, J.; Li, M. Patternhunter: Faster and more sensitive homology search. Bioinformatics 2002, 18, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Bloom, J.S.; Kruglyak, L.; Singh, M. A practical algorithm for finding maximal exact matches in large sequence datasets using sparse suffix arrays. Bioinformatics 2009, 25, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Abouelhoda, M.I.; Kurtz, S.; Ohlebusch, E. Replacing suffix trees with enhanced suffix arrays. J. Discret. Algorithms 2004, 2, 53–86. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Höhl, M.; Kurtz, S.; Ohlebusch, E. Efficient multiple genome alignment. Bioinformatics 2002, 18, S312–S320. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.; Freitas, A.T. Slamem: Efficient retrieval of maximal exact matches using a sampled lcp array. Bioinformatics 2014, 30, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Khiste, N.; Ilie, L. E-mem: Efficient computation of maximal exact matches for very large genomes. Bioinformatics 2015, 31, 509–514. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marke, L. AMAS: A fast tool for alignment manipulation and computing of summary statistics. PeerJ PrePrints 2015, 3, e1672. [Google Scholar]
- Cao, X.; Li, S.C.; Tung, A.K. Indexing DNA Sequences Using q-grams. In Database Systems for Advanced Applications; Springer: Beijing, China, 2005; pp. 4–16. [Google Scholar]
- Sankoff, D. Edit distance for genome comparison based on non-local operations. In Combinatorial Pattern Matching; Springer: Berlin, Germany, 1992; pp. 121–135. [Google Scholar]
- Ahmadi, A.; Behm, A.; Honnalli, N.; Li, C.; Weng, L.; Xie, X. Hobbes: Optimized gram-based methods for efficient read alignment. Nucleic Acids Res. 2012, 40, e41. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nature Rev. Genetics 2012, 13, 36–46. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Zheng, H.; Luo, R.; Zhu, H.; Li, Q.; Qian, W.; Ren, Y.; Tian, G.; Li, J. Building the sequence map of the human pan-genome. Nature Biotechnol. 2010, 28, 57–63. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, H.; Meehan, J.; Tong, W.; Hong, H. Alignment of Short Reads: A Crucial Step for Application of Next-Generation Sequencing Data in Precision Medicine. Pharmaceutics 2015, 7, 523-541. https://doi.org/10.3390/pharmaceutics7040523
Ye H, Meehan J, Tong W, Hong H. Alignment of Short Reads: A Crucial Step for Application of Next-Generation Sequencing Data in Precision Medicine. Pharmaceutics. 2015; 7(4):523-541. https://doi.org/10.3390/pharmaceutics7040523
Chicago/Turabian StyleYe, Hao, Joe Meehan, Weida Tong, and Huixiao Hong. 2015. "Alignment of Short Reads: A Crucial Step for Application of Next-Generation Sequencing Data in Precision Medicine" Pharmaceutics 7, no. 4: 523-541. https://doi.org/10.3390/pharmaceutics7040523
APA StyleYe, H., Meehan, J., Tong, W., & Hong, H. (2015). Alignment of Short Reads: A Crucial Step for Application of Next-Generation Sequencing Data in Precision Medicine. Pharmaceutics, 7(4), 523-541. https://doi.org/10.3390/pharmaceutics7040523