Impact of Molecular Weight on Lymphatic Drainage of a Biopolymer-Based Imaging Agent

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis of 5-Carboxypentyl-amino-IR-820
2.3. Synthesis of HA-IR820 Conjugates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HA MW (kDa) | Dye Equivalents | EDAC Equivalents | DMAP Equivalents |
|---|---|---|---|
| 6.4 | 1.5 | 1.8 | 0.75 |
| 35 | 3 | 3.6 | 1.5 |
| 74 | 10 | 12 | 5 |
| 132 | 25 | 30 | 12.5 |
| 357 | 50 | 60 | 25 |
| 697 | 100 | 120 | 50 |
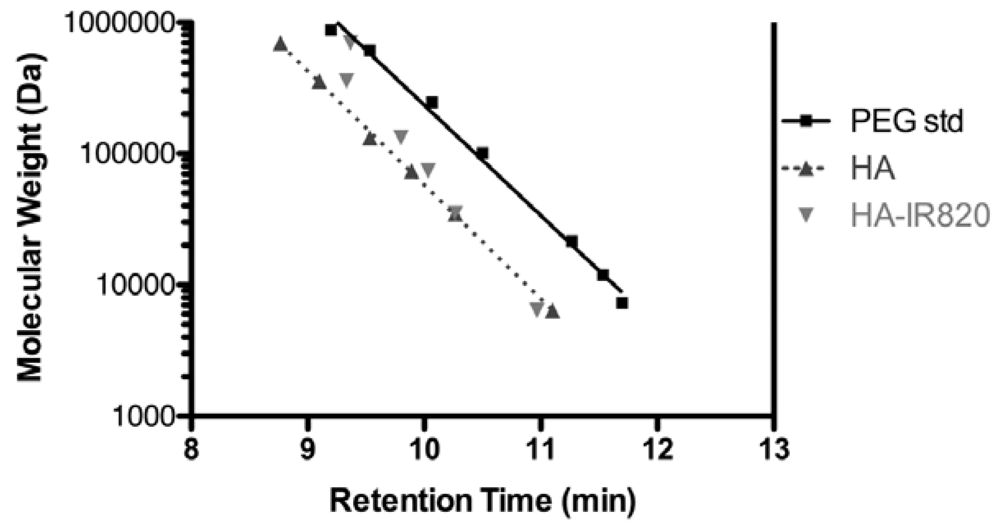
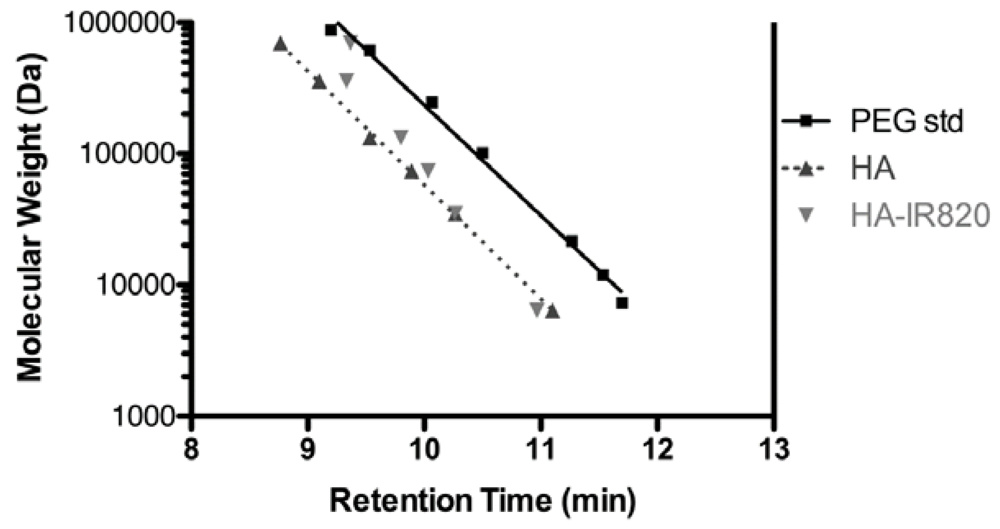
2.4. Size Exclusion Chromatography
2.5. Fluorescence
2.6. In Vivo Imaging
2.7. Data Analysis
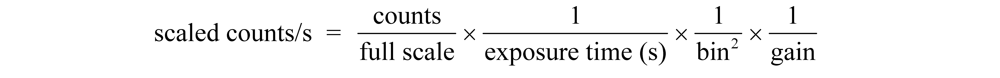
3. Results and Discussion
3.1. Choice of HA Molecular Weights
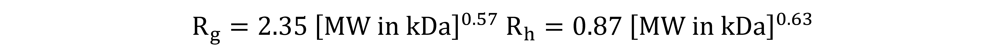
| HA MW (kDa) | Rg (nm) | Rh (nm) |
|---|---|---|
| 6.4 | 6.77 | 2.80 |
| 35 | 17.8 | 8.17 |
| 74 | 27.3 | 13.1 |
| 132 | 38.1 | 18.9 |
| 357 | 67.0 | 35.3 |
| 697 | 98.1 | 53.8 |
3.2. Polymer Characterization
| HA MW (kDa) | IR820 wt % | # Dyes/HA Polymer |
|---|---|---|
| 6.4 | 2.12% | 0.16 |
| 35 | 4.69% | 1.93 |
| 74 | 3.21% | 2.75 |
| 132 | 4.95% | 7.72 |
| 357 | 4.44% | 18.59 |
| 697 | 3.97% | 32.29 |
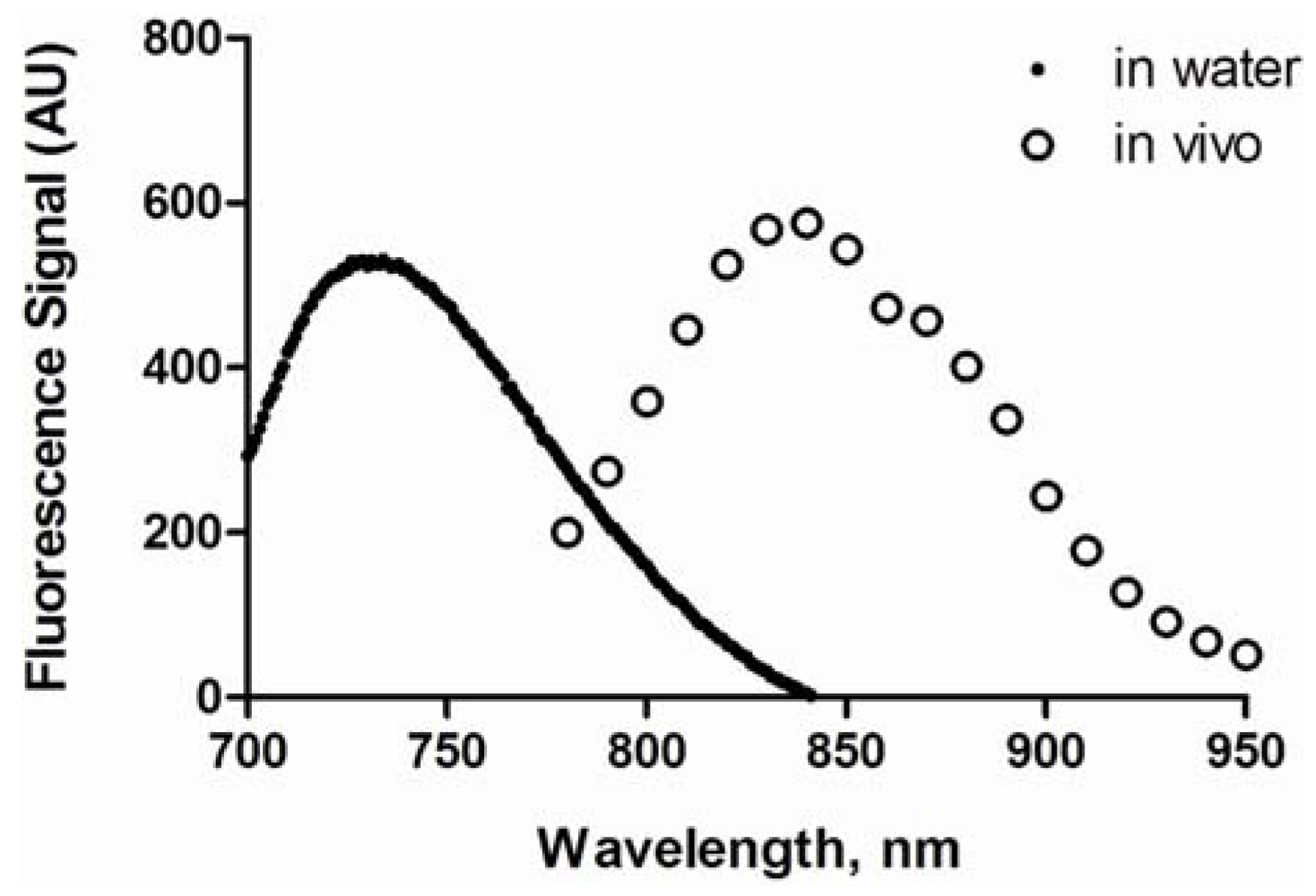
3.3. Spectral Properties
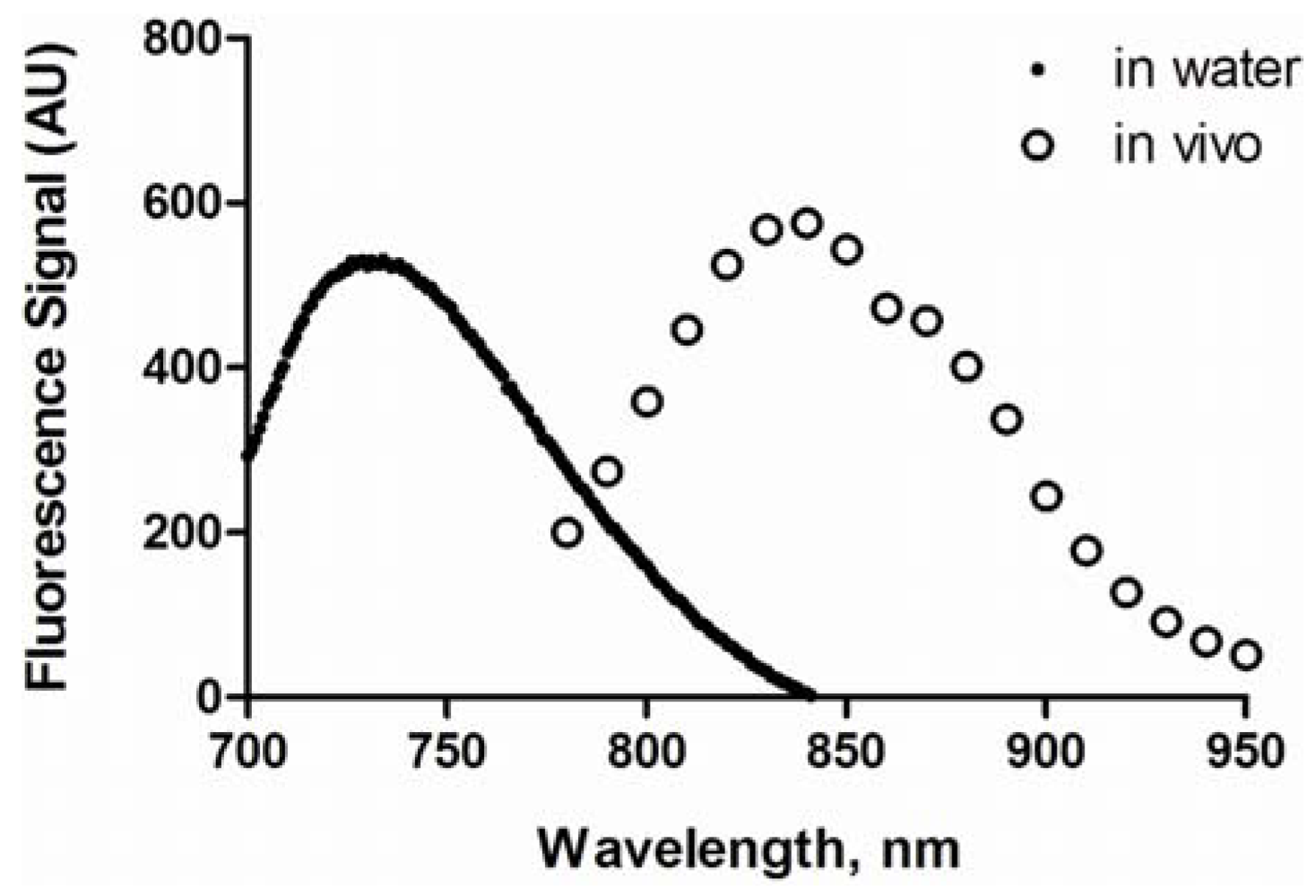
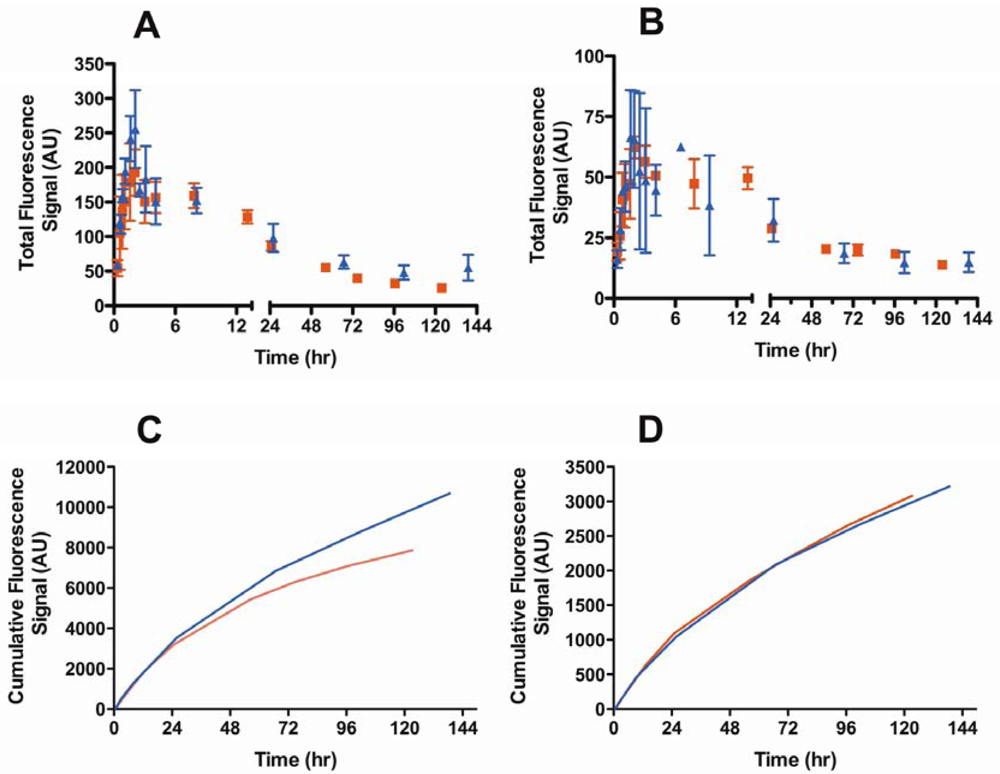
3.4. In Vivo Imaging
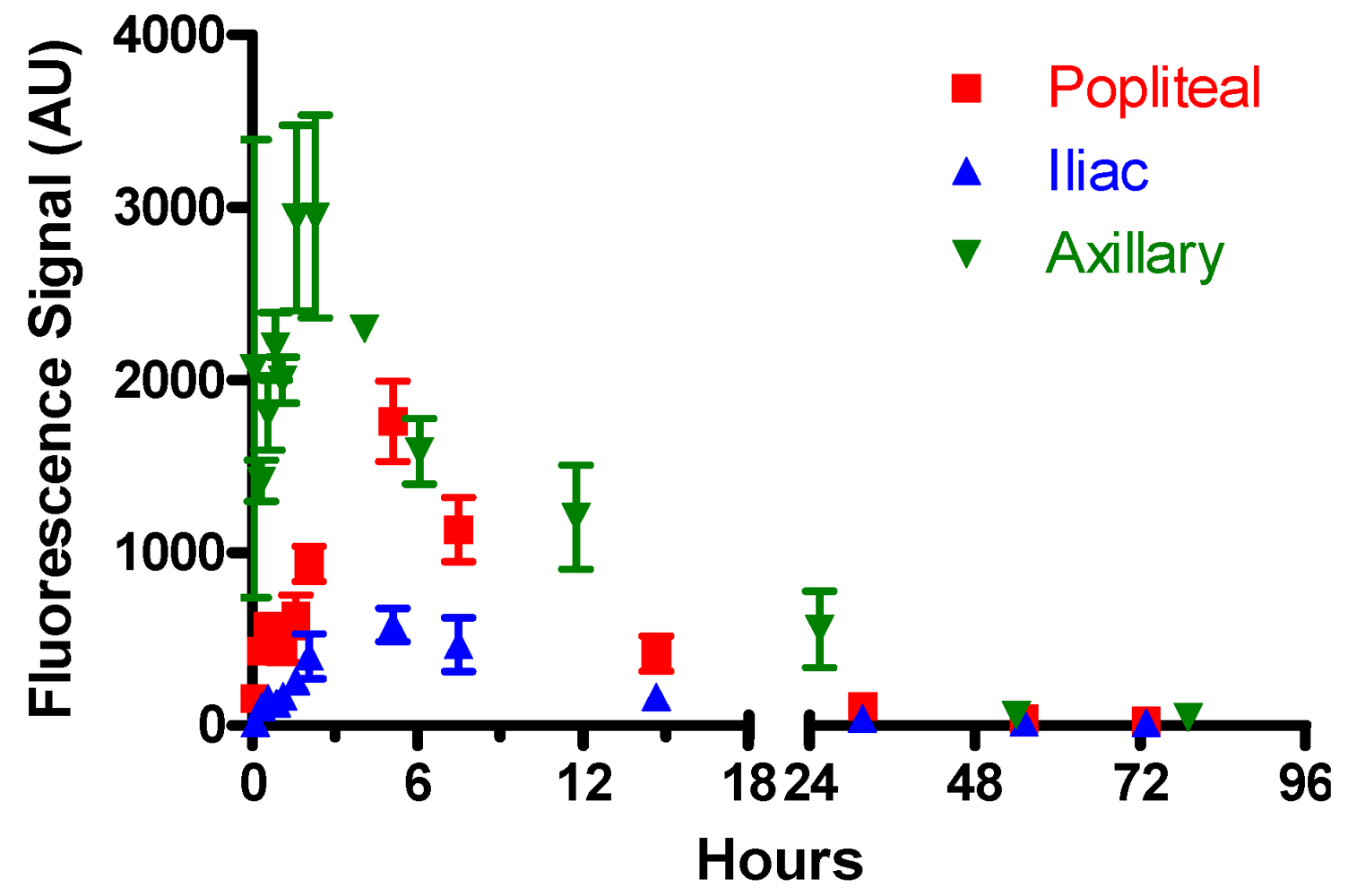
3.4.1. Optimization of Lymphatic Imaging
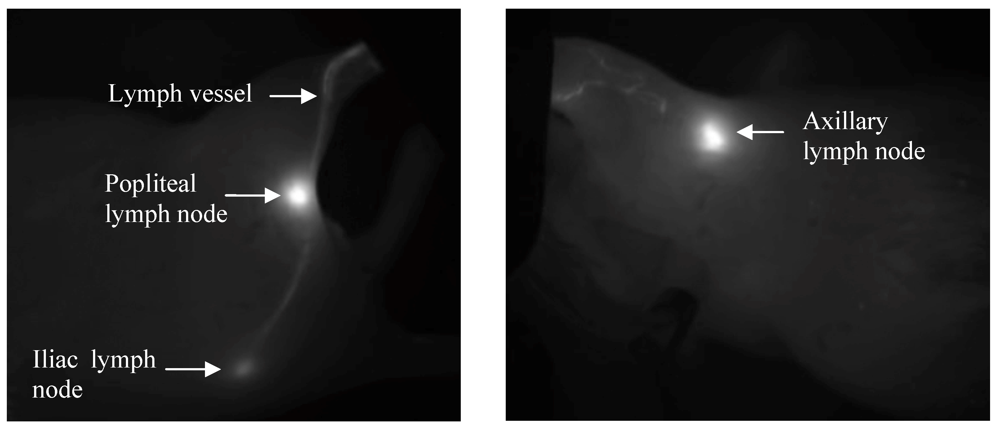
3.4.2. Control Experiments
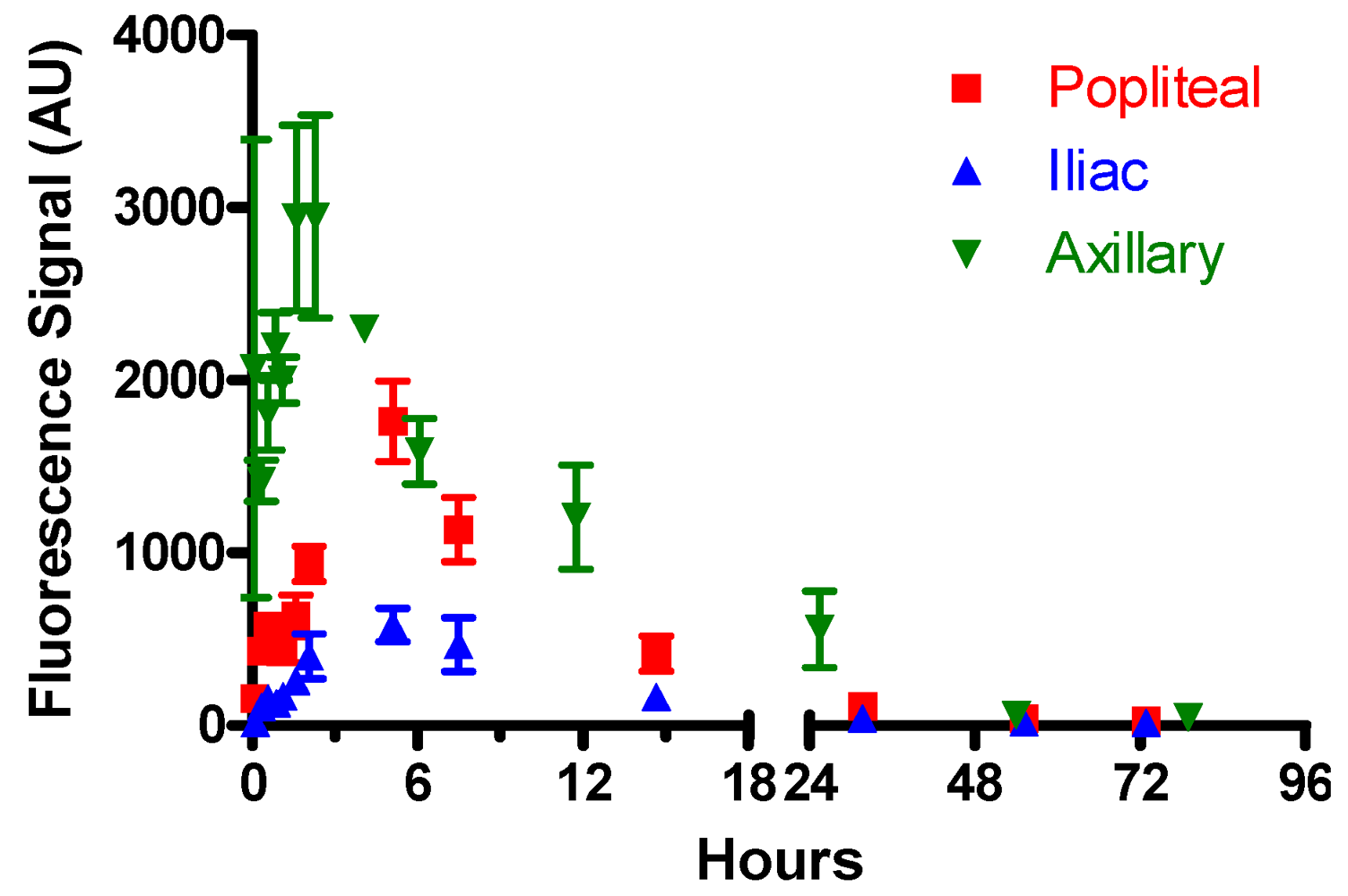
| tmax, h | t50%, h | |||||
|---|---|---|---|---|---|---|
| Sample | Popliteal | Iliac | Axillary | Popliteal | Iliac | Axillary |
| 6.4 kDa HA-IR820 | 1.5 | 4 | 1 | 7.6 ± 3.2 | 16 ± 7 | 10 ± 3 |
| 35 kDa HA-IR820 | 1.5 | 2 | 1–2 | 5.7 ± 1.4 | 4.6 ± 3.6 | 17 ± 6 |
| 74 kDa HA-IR820 | 1.5–2 | 1.5–2 | 3 | 6.9 ± 2.4 | 21 ± 1 | 15 ± 5 |
| 132 kDa HA-IR820 | 1.5 | 2–4 | 4 | 49 ± 15 | 30 ± 6 | 62 ± 22 |
| 357 kDa HA-IR820 | 3 | 3 | 3–4 | 38 ± 11 | 21 ± 9 | 62 ± 26 |
| 697 kDa HA-IR820 | 3 | 2–3 | 3 | 47 ± 9 | 62 ± 19 | 72 ± 8 |
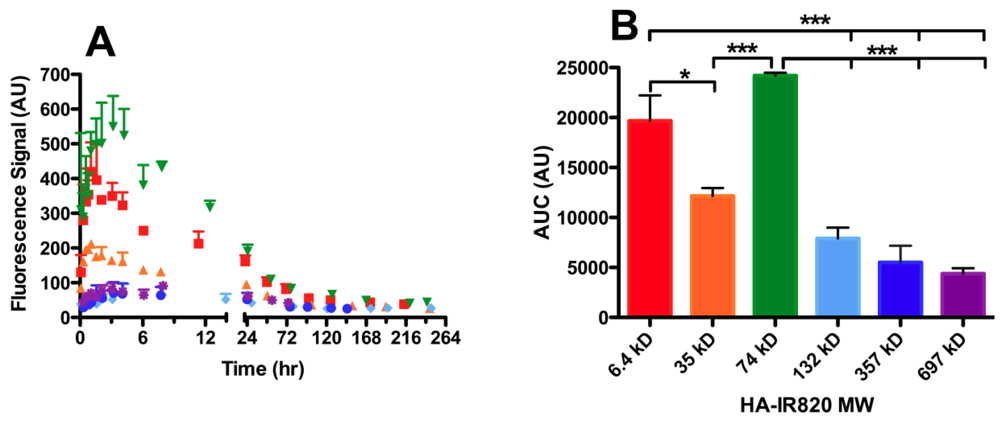
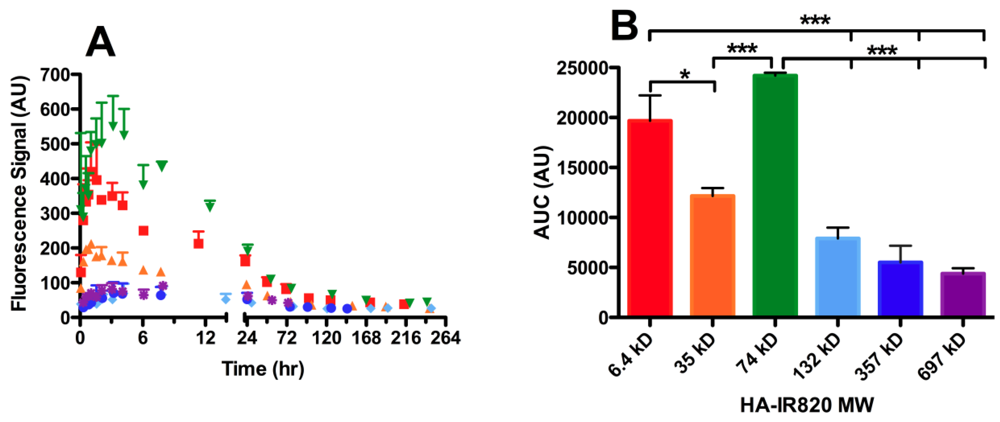
3.4.3. Molecular Weight Dependence on Lymphatic Uptake
| HA MW (kDa) | Axillary AUC (AU) | Popliteal AUC (AU) | Iliac AUC (AU) |
|---|---|---|---|
| 6.4 | 19679 ± 2530 | 9734 ± 1821 | 3860 ± 466 |
| 35 | 12155 ± 786 | 3968 ± 1512 | 1019 ± 425 |
| 74 | 24190 ± 296 | 11464 ± 1916 | 4173 ± 1071 |
| 132 | 7897 ± 1101 | 3589 ± 507 | 1505 ± 207 |
| 357 | 5514 ± 1669 | 4887 ± 385 | 1901 ± 282 |
| 697 | 4381 ± 545 | 2867 ± 469 | 1088 ± 375 |
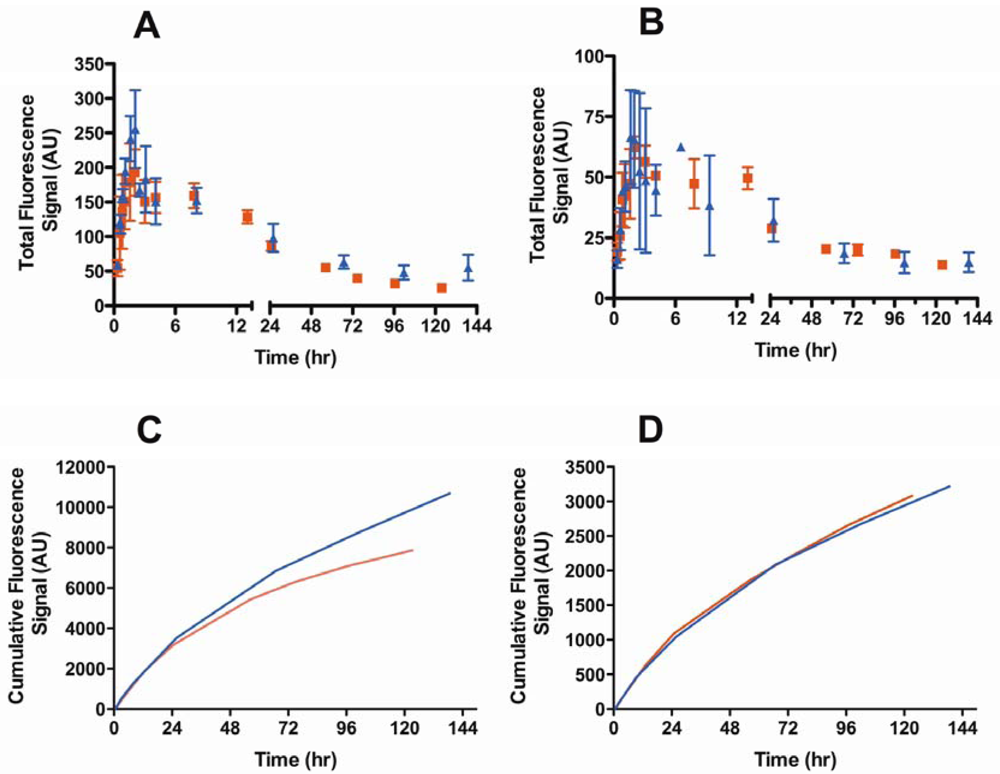
3.4.4. Lymphatic Uptake in the Presence of B16F10 Tumors
| Lymph Node | Animal | tmax (h) | t50% (h) |
|---|---|---|---|
| Popliteal | B16F10 Tumor | 2 | 20 ± 4 |
| Normal | 1.5–2 | 6.9 ± 2.4 | |
| Iliac | B16F10 Tumor | 2 | 23 ± 2 |
| Normal | 1.5–2 | 21 ± 1 |
4. Conclusions
Conflict of Interest
Acknowledgments
References
- Nathanson, S.D. Insights into the mechanisms of lymph node metastasis. Cancer 2003, 98, 413–423. [Google Scholar] [CrossRef]
- Eccles, S.A.; Welch, D.R. Metastasis: Recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.L.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar]
- White, V.; Harvey, J.R.; Griffith, C.D.; Youssef, M.; Carr, M. Sentinel lymph node biopsy in early breast cancer surgery—Working with the risks of vital blue dye to reap the benefits. Eur. J. Surg. Oncol. 2011, 37, 101–108. [Google Scholar] [CrossRef]
- Gould, E.A.; Winship, T.; Philbin, P.H.; Kerr, H.H. Observations on a “Sentinel node” in cancer of the parotid. Cancer 1960, 13, 77–78. [Google Scholar]
- Ross, M.I. Sentinel node biopsy for melanoma: An update after two decades of experience. Semin. Cutan. Med. Surg. 2011, 29, 238–248. [Google Scholar] [CrossRef]
- Lam, T.K.; Uren, R.F.; Scolyer, R.A.; Quinn, M.J.; Shannon, K.F.; Thompson, J.F. False-negative sentinel node biopsy because of obstruction of lymphatics by metastatic melanoma: The value of ultrasound in conjunction with preoperative lymphoscintigraphy. Melanoma Res. 2009, 19, 94–99. [Google Scholar] [CrossRef]
- Cochran, A.J.; Roberts, A.A.; Saida, T. The place of lymphatic mapping and sentinel node biopsy in oncology. Int. J. Clin. Oncol. 2003, 8, 139–150. [Google Scholar] [CrossRef]
- Lucarelli, R.T.; Ogawa, M.; Kosaka, N.; Turkbey, B.; Kobayashi, H.; Choyke, P.L. New approaches to lymphatic imaging. Lymphat. Res. Biol. 2009, 7, 205–214. [Google Scholar] [CrossRef]
- Hindie, E.; Groheux, D.; Brenot-Rossi, I.; Rubello, D.; Moretti, J.L.; Espie, M. The sentinel node procedure in breast cancer: Nuclear medicine as the starting point. J. Nucl. Med. 2011, 52, 405–414. [Google Scholar]
- Nune, S.K.; Gunda, P.; Majeti, B.K.; Thallapally, P.K.; Forrest, M.L. Advances in lymphatic imaging and drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 876–885. [Google Scholar] [CrossRef]
- Porter, C.J. Drug delivery to the lymphatic system. Crit. Rev. Ther. Drug Carrier Syst. 1997, 14, 333–393. [Google Scholar]
- Rao, D.A.; Forrest, M.L.; Alani, A.W.; Kwon, G.S.; Robinson, J.R. Biodegradable plga based nanoparticles for sustained regional lymphatic drug delivery. J. Pharm. Sci. 2010, 99, 2018–2031. [Google Scholar]
- Casley-Smith, J.R. The fine structure and functioning of tissue channels and lymphatics. Lymphology 1980, 13, 177–183. [Google Scholar]
- Oussoren, C.; Storm, G. Liposomes to target the lymphatics by subcutaneous administration. Adv. Drug Deliv. Rev. 2001, 50, 143–156. [Google Scholar] [CrossRef]
- Takada, M.; Hattori, S. Presence of fenestrated capillaries in the skin. Anat. Rec. 1972, 173, 213–219. [Google Scholar] [CrossRef]
- Imayama, S. Scanning and transmission electron-microscope study on the terminal blood-vessels of the rat skin. J. Invest. Dermatol. 1981, 76, 151–157. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes. Res. 2010, 2. [Google Scholar]
- Takakura, Y.; Atsumi, R.; Hashida, M.; Sezaki, H. Development of a novel polymeric prodrug of mitomycin c, mitomycin c-dextran conjugate with anionic charge. II. Disposition and pharmacokinetics following intravenous and intramuscular administration. Int. J. Pharm. 1987, 37, 145–154. [Google Scholar] [CrossRef]
- Patel, H.M.; Boodle, K.M.; Vaughan-Jones, R. Assessment of the potential uses of liposomes for lymphoscintigraphy and lymphatic drug delivery. Failure of 99m-technetium marker to represent intact liposomes in lymph nodes. Biochim. Biophys. Acta 1984, 801, 76–86. [Google Scholar] [CrossRef]
- Takakura, Y.; Hashida, M.; Sezaki, H. Lymphatic Transport after Parenteral Drug Administration. In Lymphatic Transport of Drugs; Charman, W.N., Stella, V.J., Eds.; CRC Press: Boca Raton, FL, USA, 1992; pp. 255–278. [Google Scholar]
- Hilderbrand, S.A.; Weissleder, R. Near-infrared fluorescence: Application to in vivo molecular imaging. Curr. Opin. Chem. Biol. 2010, 14, 71–79. [Google Scholar] [CrossRef]
- Kobayashi, H.; Koyama, Y.; Barrett, T.; Hama, Y.; Regino, C.A.; Shin, I.S.; Jang, B.S.; Le, N.; Paik, C.H.; Choyke, P.L.; et al. Multimodal nanoprobes for radionuclide and five-color near-infrared optical lymphatic imaging. ACS Nano 2007, 1, 258–264. [Google Scholar] [CrossRef]
- Lin, P.; Chen, J.W.; Chang, L.W.; Wu, J.P.; Redding, L.; Chang, H.; Yeh, T.K.; Yang, C.S.; Tsai, M.H.; Wang, H.J.; et al. Computational and ultrastructural toxicology of a nanoparticle, quantum dot 705, in mice. Environ. Sci. Technol. 2008, 42, 6264–6270. [Google Scholar]
- Ballou, B.; Lagerholm, B.C.; Ernst, L.A.; Bruchez, M.P.; Waggoner, A.S. Noninvasive imaging of quantum dots in mice. Bioconjug. Chem. 2004, 15, 79–86. [Google Scholar]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Itty Ipe, B.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef]
- Ossipov, D.A. Nanostructuredhyaluronic acid-based materials for active delivery to cancer. Expert Opin. Drug Deliv. 2010, 7, 681–703. [Google Scholar] [CrossRef]
- Luo, Y.; Ziebell, M.R.; Prestwich, G.D. A hyaluronic acid-taxol antitumor bioconjugate targeted to cancer cells. Biomacromolecules 2000, 1, 208–218. [Google Scholar] [CrossRef]
- Platt, V.M.; Szoka, F.C., Jr. Anticancer therapeutics: Targeting macromolecules and nanocarriers to hyaluronan or cd44, a hyaluronan receptor. Mol. Pharm. 2008, 5, 474–486. [Google Scholar] [CrossRef]
- Gaffney, J.; Matou-Nasri, S.; Grau-Olivares, M.; Slevin, M. Therapeutic applications of hyaluronan. Mol. Biosyst. 2010, 6, 437–443. [Google Scholar] [CrossRef]
- Cai, S.; Xie, Y.; Davies, N.M.; Cohen, M.S.; Forrest, M.L. Pharmacokinetics and disposition of a localized lymphatic polymeric hyaluronan conjugate of cisplatin in rodents. J. Pharm. Sci. 2010, 99, 2664–2671. [Google Scholar]
- Cai, S.; Xie, Y.; Davies, N.M.; Cohen, M.S.; Forrest, M.L. Carrier-based intralymphaticcisplatin chemotherapy for the treatment of metastatic squamous cell carcinoma of the head & neck. Ther. Deliv. 2010, 1, 237–245. [Google Scholar] [CrossRef]
- Cai, S.; Thati, S.; Bagby, T.R.; Diab, H.M.; Davies, N.M.; Cohen, M.S.; Forrest, M.L. Localized doxorubicin chemotherapy with a biopolymericnanocarrier improves survival and reduces toxicity in xenografts of human breast cancer. J. Control. Release 2010, 146, 212–218. [Google Scholar] [CrossRef]
- Cai, S.; Xie, Y.; Bagby, T.R.; Cohen, M.S.; Forrest, M.L. Intralymphatic chemotherapy using a hyaluronan-cisplatin conjugate. J. Surg. Res. 2008, 147, 247–252. [Google Scholar] [CrossRef]
- Cohen, M.S.; Cai, S.; Xie, Y.; Forrest, M.L. A novel intralymphaticnanocarrier delivery system for cisplatin therapy in breast cancer with improved tumor efficacy and lower systemic toxicity in vivo. Am. J. Surg. 2009, 198, 781–786. [Google Scholar] [CrossRef]
- Xie, Y.; Aillon, K.L.; Cai, S.; Christian, J.M.; Davies, N.M.; Berkland, C.J.; Forrest, M.L. Pulmonary delivery of cisplatin-hyaluronan conjugates via endotracheal instillation for the treatment of lung cancer. Int. J. Pharm. 2010, 392, 156–163. [Google Scholar] [CrossRef]
- Takahashi, R.; Kubota, K.; Kawada, M.; Okamoto, A. Effect of molecular weight distribution on the solution properties of sodium hyaluronate in 0.2 M NaCl solution. Biopolymers 1999, 50, 87–98. [Google Scholar] [CrossRef]
- Masotti, A.; Vicennati, P.; Boschi, F.; Calderan, L.; Sbarbati, A.; Ortaggi, G. A novel near-infrared indocyanine dye-polyethylenimine conjugate allows DNA delivery imaging in vivo. Bioconjug. Chem. 2008, 19, 983–987. [Google Scholar] [CrossRef]
- Lapcik, L.J.; Lapcik, L.; de Smedt, S.; Demeester, J.; Chabrecek, P. Hyaluronan: Preparation, structure, properties, and applications. Chem. Rev. 1998, 98, 2663–2684. [Google Scholar] [CrossRef]
- Devanand, K.; Selser, J.C. Asymptotic behavior and long-range interactions in aqueous solutions of poly(ethylene oxide). Macromolecules 1991, 24, 5943–5947. [Google Scholar] [CrossRef]
- Young, J.J.; Cheng, K.M.; Tsou, T.L.; Liu, H.W.; Wang, H.J. Preparation of cross-linked hyaluronic acid film using 2-chloro-1-methylpyridinium iodide or water-soluble 1-ethyl-(3,3-dimethylaminopropyl)carbodiimide. J. Biomater. Sci. Polym. Ed. 2004, 15, 767–780. [Google Scholar] [CrossRef]
- Birkinshaw, C.; Collins, M.N. Comparison of the effectiveness of four different crosslinking agents with hyaluronic acid hydrogel films for tisslue-culture applications. J. Appl. Polym. Sci. 2007, 104, 3183–3191. [Google Scholar] [CrossRef]
- Tilney, N.L. Patterns of lymphatic drainage in the adult laboratory rat. J. Anat. 1971, 109, 369–383. [Google Scholar]
- Hellman, S.; Harris, J.R. The appropriate breast cancer paradigm. Cancer Res. 1987, 47, 339–342. [Google Scholar]
- Uren, R.F.; Howman-Giles, R.; Thompson, J.F. Patterns of lymphatic drainage from the skin in patients with melanoma. J. Nucl. Med. 2003, 44, 570–582. [Google Scholar]
- Shimazu, K.; Noguchi, S. Sentinel lymph node biopsy before versus after neoadjuvant chemotherapy for breast cancer. Surg. Today 2011, 41, 311–316. [Google Scholar] [CrossRef]
- Ruddell, A.; Harrell, M.I.; Minoshima, S.; Maravilla, K.R.; Iritani, B.M.; White, S.W.; Partridge, S.C. Dynamic contrast-enhanced magnetic resonance imaging of tumor-induced lymph flow. Neoplasia 2008, 10, 706–713. [Google Scholar]
- Proulx, S.T.; Luciani, P.; Derzsi, S.; Rinderknecht, M.; Mumprecht, V.; Leroux, J.C.; Detmar, M. Quantitative imaging of lymphatic function with liposomal indocyanine green. Cancer Res. 2010, 70, 7053–7062. [Google Scholar]
- Lymphazurin (isosulfan blue) information from drugs.Com. Available online: http://www.drugs.com/pro/lymphazurin.html (accessed on 4 September 2011).
- West, D.C.; Hampson, I.N.; Arnold, F.; Kumar, S. Angiogenesis induced by degradation products of hyaluronic acid. Science 1985, 228, 1324–1326. [Google Scholar]
- Wolny, P.M.; Banerji, S.; Gounou, C.; Brisson, A.R.; Day, A.J.; Jackson, D.G.; Richter, R.P. Analysis of cd44-hyaluronan interactions in an artificial membrane system: Insights into the distinct binding properties of high and low molecular weight hyaluronan. J. Biol. Chem. 2010, 285, 30170–30180. [Google Scholar]
- Bot, P.T.; Pasterkamp, G.; Goumans, M.J.; Strijder, C.; Moll, F.L.; de Vries, J.P.; Pals, S.T.; de Kleijn, D.P.; Piek, J.J.; Hoefer, I.E. Hyaluronic acid metabolism is increased in unstable plaques. Eur. J. Clin. Invest. 2010, 40, 818–827. [Google Scholar] [CrossRef]
- Oussoren, C.; Zuidema, J.; Crommelin, D.J.; Storm, G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid compostion and lipid dose. Biochim. Biophys. Acta 1997, 1328, 261–272. [Google Scholar] [CrossRef]
- Zubris, K.A.; Khullar, O.V.; Griset, A.P.; Gibbs-Strauss, S.; Frangioni, J.V.; Colson, Y.L.; Grinstaff, M.W. Ease of synthesis, controllable sizes, and in vivo large-animal-lymph migration of polymeric nanoparticles. ChemMedChem 2010, 5, 1435–1438. [Google Scholar] [CrossRef]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Khullar, O.V.; Griset, A.P.; Gibbs-Strauss, S.L.; Chirieac, L.R.; Zubris, K.A.; Frangioni, J.V.; Grinstaff, M.W.; Colson, Y.L. Nanoparticle migration and delivery of paclitaxel to regional lymph nodes in a large animal model. J. Am. Coll. Surg. 2012, 214, 328–337. [Google Scholar] [CrossRef]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [Google Scholar] [CrossRef]
- McLennan, D.N.; Porter, C.J.H.; Charman, S.A. Subcutaneous drug delivery and the role of the lymphatics. Drug Discov. Today Technol. 2005, 2, 89–96. [Google Scholar] [CrossRef]
- McLennan, D.N.; Porter, C.J.; Edwards, G.A.; Martin, S.W.; Charman, S.A. Molecular weight is a primary determinant for lymphatic absorption of proteins following subcutaneous administration to sheep. AAPS PharmSci 2002, 4, W4041. [Google Scholar]
- Reddy, S.T.; Berk, D.A.; Jain, R.K.; Swartz, M.A. A sensitive in vivo model for quantifying interstitial convective transport of injected macromolecules and nanoparticles. J. Appl. Physiol. 2006, 101, 1162–1169. [Google Scholar] [CrossRef]
- Harrell, M.I.; Iritani, B.M.; Ruddell, A. Tumor-induced sentinel lymph node lymphangiogenesis and increased lymph flow precede melanoma metastasis. Am. J. Pathol. 2007, 170, 774–786. [Google Scholar] [CrossRef]
- Warso, M.; Gray, T.; Gonzalez, M. Melanoma of the hand. J. Hand Surg. Am. 1997, 22, 354–360. [Google Scholar] [CrossRef]
- Liu, N.F. Trafficking of hyaluronan in the interstitium and its possible implications. Lymphology 2004, 37, 6–14. [Google Scholar]
- Kobayashi, H.; Kawamoto, S.; Bernardo, M.; Brechbiel, M.W.; Knopp, M.V.; Choyke, P.L. Delivery of gadolinium-labeled nanoparticles to the sentinel lymph node: Comparison of the sentinel node visualization and estimations of intra-nodal gadolinium concentration by the magnetic resonance imaging. J. Control. Release 2006, 111, 343–351. [Google Scholar] [CrossRef]
- Kaminskas, L.M.; Kota, J.; McLeod, V.M.; Kelly, B.D.; Karellas, P.; Porter, C.J. Pegylation of polylysinedendrimers improves absorption and lymphatic targeting following sc administration in rats. J. Control. Release 2009, 140, 108–116. [Google Scholar] [CrossRef]
- Supersaxo, A.; Hein, W.R.; Steffen, H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm. Res. 1990, 7, 167–169. [Google Scholar] [CrossRef]
- Wijagkanalan, W.; Kawakami, S.; Hashida, M. Designing dendrimers for drug delivery and imaging: Pharmacokinetic considerations. Pharm. Res. 2011, 28, 1500–1519. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bagby, T.R.; Cai, S.; Duan, S.; Thati, S.; Aires, D.J.; Forrest, L. Impact of Molecular Weight on Lymphatic Drainage of a Biopolymer-Based Imaging Agent. Pharmaceutics 2012, 4, 276-295. https://doi.org/10.3390/pharmaceutics4020276
Bagby TR, Cai S, Duan S, Thati S, Aires DJ, Forrest L. Impact of Molecular Weight on Lymphatic Drainage of a Biopolymer-Based Imaging Agent. Pharmaceutics. 2012; 4(2):276-295. https://doi.org/10.3390/pharmaceutics4020276
Chicago/Turabian StyleBagby, Taryn R., Shuang Cai, Shaofeng Duan, Sharadvi Thati, Daniel J. Aires, and Laird Forrest. 2012. "Impact of Molecular Weight on Lymphatic Drainage of a Biopolymer-Based Imaging Agent" Pharmaceutics 4, no. 2: 276-295. https://doi.org/10.3390/pharmaceutics4020276
APA StyleBagby, T. R., Cai, S., Duan, S., Thati, S., Aires, D. J., & Forrest, L. (2012). Impact of Molecular Weight on Lymphatic Drainage of a Biopolymer-Based Imaging Agent. Pharmaceutics, 4(2), 276-295. https://doi.org/10.3390/pharmaceutics4020276