1. Introduction
Paclitaxel (PTX) is a diterpenoid alkaloid compound with significant anticancer activity, first isolated from the bark of short-leafed redwood (
Taxus brevifolia Nutt.) in 1971 by the research team of Wall and Wani. Its pharmacological mechanism of action is mainly through the induction and promotion of microtubule protein polymerisation, microtubule assembly, and the prevention of depolymerisation, thus stabilising the microtubule: paclitaxel specifically binds to β-microtubulin subunits, alters the dynamic equilibrium between soluble microtubule proteins and polymers, and significantly reduces the critical concentration of microtubule assembly, thus leading to cell cycle arrest in the G2/M phase [
1]. Based on its unique anti-tumour mechanism and broad-spectrum anti-tumour activity, this drug has been widely used clinically in the treatment of solid tumours such as breast cancer, ovarian cancer and non-small-cell lung cancer [
2]. However, PTX faces two major challenges for clinical use: First, its very low water solubility (0.1 μg/mL) makes formulation development difficult. Currently, Cremophor EL and anhydrous ethanol (1:1,
v/
v) are commonly used as solubilising carriers and should be diluted to 0.3–1.2 mg/mL in saline or 5% dextrose solution and then infused intravenously [
3]. Second, there are significant safety concerns with this excipient system: studies have demonstrated that Cremophor EL induces severe anaphylactoid reactions (30–40% incidence), possibly related to complement activation and histamine release, as well as dose-dependent neurotoxicity, erythrocyte aggregation, and lipid metabolism disorders [
4]. Although allergic reactions can be partially alleviated by premedication with antihistamines and glucocorticoids, the resulting complexity of the medication regimen significantly reduces patient compliance [
5]. To overcome these limitations, the development of novel drug delivery systems has become a hot research topic. Two improved formulations have already gained clinical acceptance: liposome-encapsulated formulations to improve drug solubility through phospholipid bilayers and albumin-conjugated nanoparticles (Abraxane
®, Celgene, Samet, NJ, USA) to achieve targeted delivery using endogenous transport proteins [
6,
7]. Although these new dosage forms can significantly reduce the incidence of adverse reactions, there are still industrialisation problems such as their complex preparation process and high production cost, which, to a certain extent, limits their clinical promotion and application.
Solutol HS-15 is a high-performance amphiphilic nonionic surfactant that has been extensively studied for its excellent solubilising ability, low haemolytic activity and good biocompatibility [
8]. Solutol HS-15 offers significant advantages over conventional Cremophor EL: it not only enables low volume injections of high-dose drugs, thereby reducing solvent-related toxicity, but it also significantly improves patient compliance by eliminating the need for the premedication of antihistamines or corticosteroids due to its low immunogenicity. In addition, Solutol HS-15 reverses multidrug resistance (MDR) in tumour cells by inhibiting the efflux function of P-glycoprotein (P-gp) [
9], which is particularly important for the intracellular accumulation of strong P-gp substrates such as PTX [
10]. Therefore, Solutol HS-15 is considered a highly promising paclitaxel delivery vehicle, which is expected to replace the conventional Cremophor EL-based formulation.
Glioma, the most aggressive primary malignant tumour of the central nervous system, has an extremely poor clinical prognosis with a 5-year survival rate of less than 5%. Despite the use of multimodality treatment, including aggressive surgical resection combined with radiotherapy and/or chemotherapy, the prognosis for patients with gliomas is still very poor [
11]. It is mainly limited by two major factors: firstly, the strict selectivity of the blood–brain barrier (BBB), which makes it difficult for systemically administered chemotherapeutic drugs to reach effective concentrations at the tumour site, and secondly, resistance due to tumour heterogeneity and drug efflux mechanisms [
12]. In recent years, advances in nanotechnology have provided new ideas to overcome the BBB, such as the modification of nanocarriers by targeted ligands such as transferrin and Angiopep-2, or the use of cell-penetrating peptide (CPP) and stimulus-responsive (e.g., pH/enzyme-sensitive) nanosystems to enhance drug delivery efficiency [
13]. In addition to chemical materials, biocarriers are also at the forefront. Glioma cell-derived exosomes or extracellular vesicles containing characteristic proteins on their surfaces are ideal targeted delivery vectors for therapeutic studies of gliomas [
14], and surface remodelling with functional groups can further enhance their performance, like chimeric antigen receptors or oligopeptides [
15,
16]. However, the clinical translation of these complex nanosystems still faces significant challenges, mainly stemming from the complexity of their preparation process, difficulty in scale-up production, and inconsistent efficacy due to poor batch-to-batch consistency. Therefore, the development of nanoformulations with simple, efficient and easy-to-industrialise processes is a key direction to drive therapeutic breakthroughs in glioma treatment.
In addition to active targeted ligand modification, passively targeted nanoparticles based on blood–brain–tumour barrier (BBTB) leakage properties and enhanced permeation retention (EPR) effects provide a facile and effective delivery strategy. In the physiological state, substances enter the brain tissue mainly via the paracellular pathway and passive diffusion, but the tight junctions between endothelial cells limit the transport of most small molecules and all macromolecules across the barrier [
17]. This method is therefore inappropriate under normal physiological conditions. However, in the pathological state of gliomas, tumour infiltration leads to the destruction of the BBB structure and the formation of the BBTB [
18]. In this context, the BBTB becomes a major barrier to anticancer drug delivery. With tumour progression, BBTB integrity is further impaired, as evidenced by the increased vascular permeability and disruption of tight junctions, a pathological feature that creates conditions for EPR effects [
19]. It has been shown that the down-regulation of the expression of the tight junction-specific protein Claudin-1 in the peritumoural vasculature is one of the key molecular mechanisms leading to increased BBTB permeability [
20]. Under these conditions, the particle size of the nanocarrier becomes a decisive factor influencing the delivery efficiency: small-sized nanoparticles (<20 nm) not only enhance brain tumour targeting but also reduce hepatic uptake, which reduces the first-pass effect and improves the therapeutic index [
21]. These findings suggest that untargeted nano-formulations with optimised particle size may achieve glioma-specific accumulation and have better clinical translational potential.
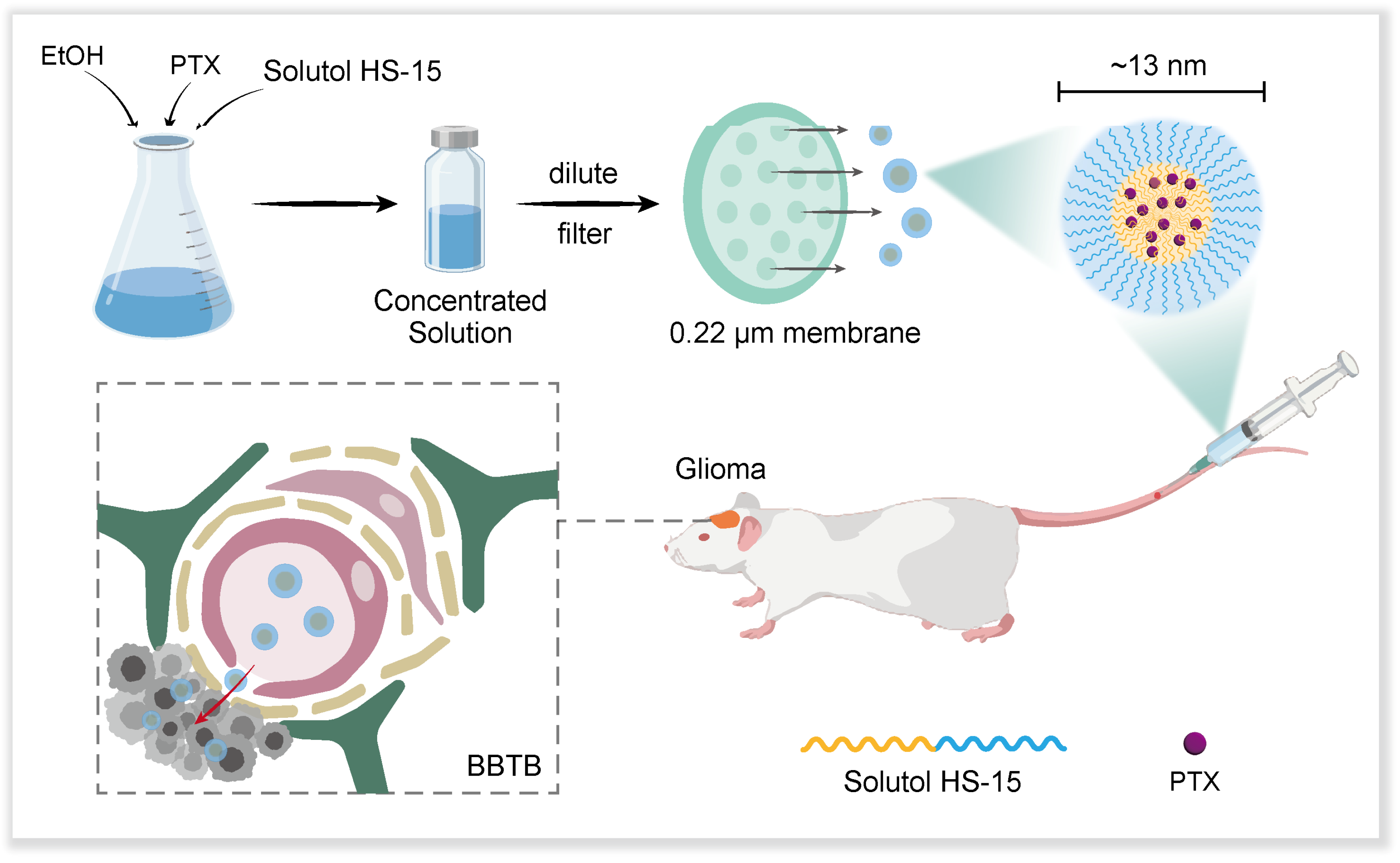
Based on the above theoretical foundation, our research team innovatively developed a novel paclitaxel nano-formulation using Solutol HS-15 as the carrier material (
Scheme 1). The formulation has the following significant advantages: (1) the preparation process is simple, and only saline dilution is required to form drug-carrying micelles with a particle size of about 13 nm; (2) it exhibits excellent tumour targeting in an in situ mouse model of glioma; (3) it has significantly reduced systemic toxicity compared with the traditional Taxol formulation; and (4) it exhibits significant anti-tumour efficacy. This nano-formulation based on passive targeting strategy provides a new R&D idea for the clinical treatment of glioma, and its simple preparation process and good therapeutic effect show outstanding translational medical value.
2. Materials and Methods
2.1. Materials, Cells and Animals
Ethanol, tween-80, and cell-grade DMSO were purchased from Sigma-Aldrich Inc. (Shanghai, China). PTX was purchased from Beijing Kai Guo Technology Co. (Beijing, China). Solutol HS-15 was kindly donated by Beijing Fengli Jingqiu Pharmaceutical Co. (Beijing, China), DAPI was purchased from Beyotime Institute of Biotechnology (Shanghai, China), Cell Counting Kit-8 (CCK-8) was purchased from Solarbio Ltd. (Beijing, China), Foetal Bovine Serum (FBS), Trypsin, 1640 medium, and PBS were purchased from Thermo Fisher Scientific Co., Ltd. (Beijing, China), and the DiR fluorescent probe was purchased from Solarbio Ltd. (Beijing, China). FITC-PTX was purchased from New Weichuang Biotechnology (Chongqing) Co., (Chongqing, China), and BUN, ALT, AST, and CRE test kits were purchased from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Aphrodite (ready-to-use tribromoethanol) was purchased from Melunun Cell Biotechnology Co. (Beijing, China). D-Luciferin (potassium salt) was purchased from APExBIO Technology LLC (Shanghai, China). Universal Tissue Fixative and 4% paraformaldehyde fixative were purchased from Wuhan Servicebio Technology Co. (Wuhan, China), and the Taxol-positive model drug was prepared by this experiment according to the public prescription of listed drugs. C6 cells were purchased from the Department of Pathology, the Institute of Pharmaceutical Biotechnology, Peking Union Medical College, Peking, China, and a stable luciferase-transfected cell line (C6Luc) was constructed in our laboratory. They were grown in RPMI 1640 medium containing 10% FBS at 37 °C in 5% CO2 atmosphere. Female BALB/c mice (5–8 weeks old, 18–22 g) were purchased from Beijing GemPharmatech Co. (Beijing, China). All animal studies were approved by the Laboratory Animal Ethics Committee of the Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC) under the number 00004445. All experimental procedures were conducted in accordance with institutional guidelines and protocols for the care and use of experimental animals.
2.2. Preparation and Characterisation of PSMs
Solutol HS-15 and paclitaxel (30:1, w/w) were weighed and dissolved in a certain amount of ethanol and ultrasonicated (power 300 W and frequency 40 kHz) for 10 min until completely dissolved to produce a concentrated solution 1 of 12 mg/mL; another set of Solutol HS-15 and paclitaxel (60:1, w/w) was weighed and dissolved in a certain amount of ethanol to produced concentrated solution 2 of 6 mg/mL by the same method. The two concentrated solutions were stored at 4 °C, protected from light, diluted to 1 mg/mL with double-distilled water or saline before use, and filtered through 0.22 μm microporous membrane (Millipore, New Bedford, MA, USA). Blank micelles (SMs) were prepared with reference to the process parameters of concentrated solution 2, only without the addition of PTX.
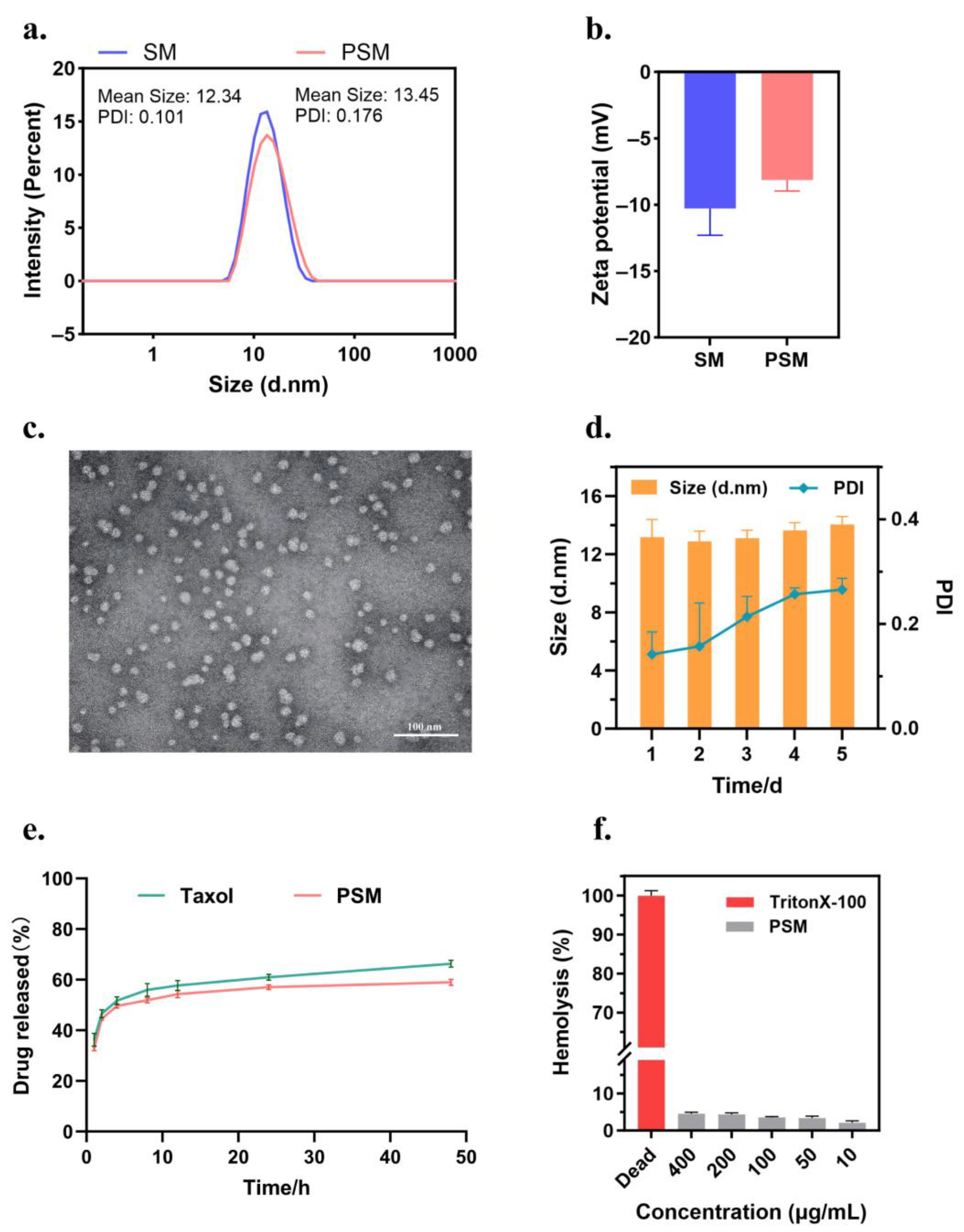
The hydrodynamic diameter, polydispersity index (PDI) and zeta potential of the nanocolloid micelles were determined by dynamic light scattering (DLS) at 25 °C (Malvern Zetasizer Nano ZS90, Malvern instruments Ltd., Worcestershire, UK); the medium was purified water, and purified water was used for dilution. To verify the morphological characteristics of the formulations, the optimal prescription samples were selected, diluted and dripped on a copper grid, negatively stained with phosphotungstic acid, and then subjected to natural evaporation to immobilise the staining agent, followed by transmission electron microscopy (TEM, Hitachi H-7650, Hitachi Ltd., Tokyo, Japan) at an accelerating voltage of 80 kV. In addition, the in vitro stability of the formulations was evaluated by continuously monitoring the changes in particle size and the PDI over 5 days. Three parallel samples were set up for all experiments (n = 3).
The HPLC determination of drug loading (DL) and encapsulation (EE) in PSM: A portion of the diluted sample was filtered through a 0.22 µm membrane to remove free PTX and used to determine the concentration of PTX in the micelles; the unfiltered sample was used to determine the feeding concentration of PTX. The total weight of the micelles was the sum of the weight of the carrier material (Solutol HS-15) and the weight of PTX encapsulated within. DL (%) and EE (%) were calculated by the following equations: EE (%) = (weight of PTX in micelle/feeding weight of PTX) × 100%, DL (%) = (weight of PTX in micelle/weight of the micelle) × 100%. PTX concentrations were detected by an Agilent 1200 LC (Agilent Tech, Santa Clara, CA, USA) high-performance liquid chromatography system using an Inertsustain C18 column (5 µm, 4.6 mm × 250 mm). The mobile phase was acetonitrile and water (60:40, v/v) at a flow rate of 1.0 mL/min. The injection volume was 20 µL, the temperature was 25 °C, and the wavelength was set at 227 nm.
The in vitro drug release assay of PSMs: The concentrated solutions of Taxol and PSMs were diluted to a concentration of 1 mg/mL of PTX, filtered through a 0.22 µm microporous membrane, and then determined by HPLC to obtain the specific concentration. In total, 1 mL of each solution was placed in a dialysis bag (MWCO = 12 kDa), tied at both ends with a thin string, and immersed in 40 mL of PBS (0.5% Tween 80, pH 7.4) at 100 rpm, 37 °C. A volume of 1 mL of release medium was sampled at predetermined time intervals (1, 2, 4, 8, 12, 24, and 48 h) and then immediately replaced with an equal amount of fresh PBS. The collected medium was filtered through a 0.22 µm microporous filter membrane and the different samples were analysed by HPLC (n = 3). The cumulative drug release concentration was calculated by the following equation: Cumulative release (%) = (V0 × Ct + V × ∑Ci)/M × 100%. V0 is the total volume of the release medium (40 mL), Ct is the concentration of the drug measured at the last sampling point, V is the volume of each sample (1 mL), ∑Ci is the concentration of the drug measured at all sampling moments prior to the first to the last sampling point, and M is the total amount of drug in the formulation.
The haemolytic assay of PSM: Healthy murine blood was taken and red blood cell suspension was prepared. Specifically, mouse blood was removed from fibrinogen and added to 0.9% sodium chloride solution, shaken well and centrifuged to remove the supernatant, and the precipitated erythrocytes were continued to be washed several times until the supernatant did not show a distinct red colour. The PSM preparation was prepared corresponding to PTX concentrations of 400, 200, 100, 50, and 10 μg/mL, and TritonX-100 (Sigma-Aldrich Inc., Shanghai, China) was used as a positive control, and PBS was used as a negative control. The samples were co-incubated with the erythrocyte suspensions at 37 °C and then centrifuged, and the supernatants were taken, and the supernatant was recorded using an enzyme marker (BioTek, Dallas, TX, USA) at 540 nm. OD values and the haemolysis rate were calculated as follows: Hemolysis rate (%) = [(ODsample − ODnegative)/(ODpositive − ODnegative)] × 100%.
Critical micelle concentration (CMC) determination: Critical micelle concentration was determined using a pyrene fluorescent probe. Specifically, the acetone solution of pyrene was prepared and added to 10 volumetric flasks, and after the acetone evaporated to dryness, micellar solution was added and fixed with purified water so that the micellar concentration was 5 × 10−4, 1 × 10−3, 5 × 10−3, 1 × 10−2, 5 × 10−2, 1 × 10−1, 5 × 10−1, 1, 5, and 10 mg/mL and then left to stand for 24 h after appropriate sonication. Determination was carried out using a fluorescence spectrophotometer, with an excitation wavelength of 336 nm, an emission wavelength scanning range of 350–450 nm, a slit width of 5 nm, and a medium scanning speed, and the fluorescence intensity of each sample, I1 and I3, was recorded sequentially. The concentration I1/I3 ratio was used to make a fitting curve, and the inflexion point of the curve (the intersection of two straight line segments) was the CMC.
2.3. In Vitro Cell Experiment
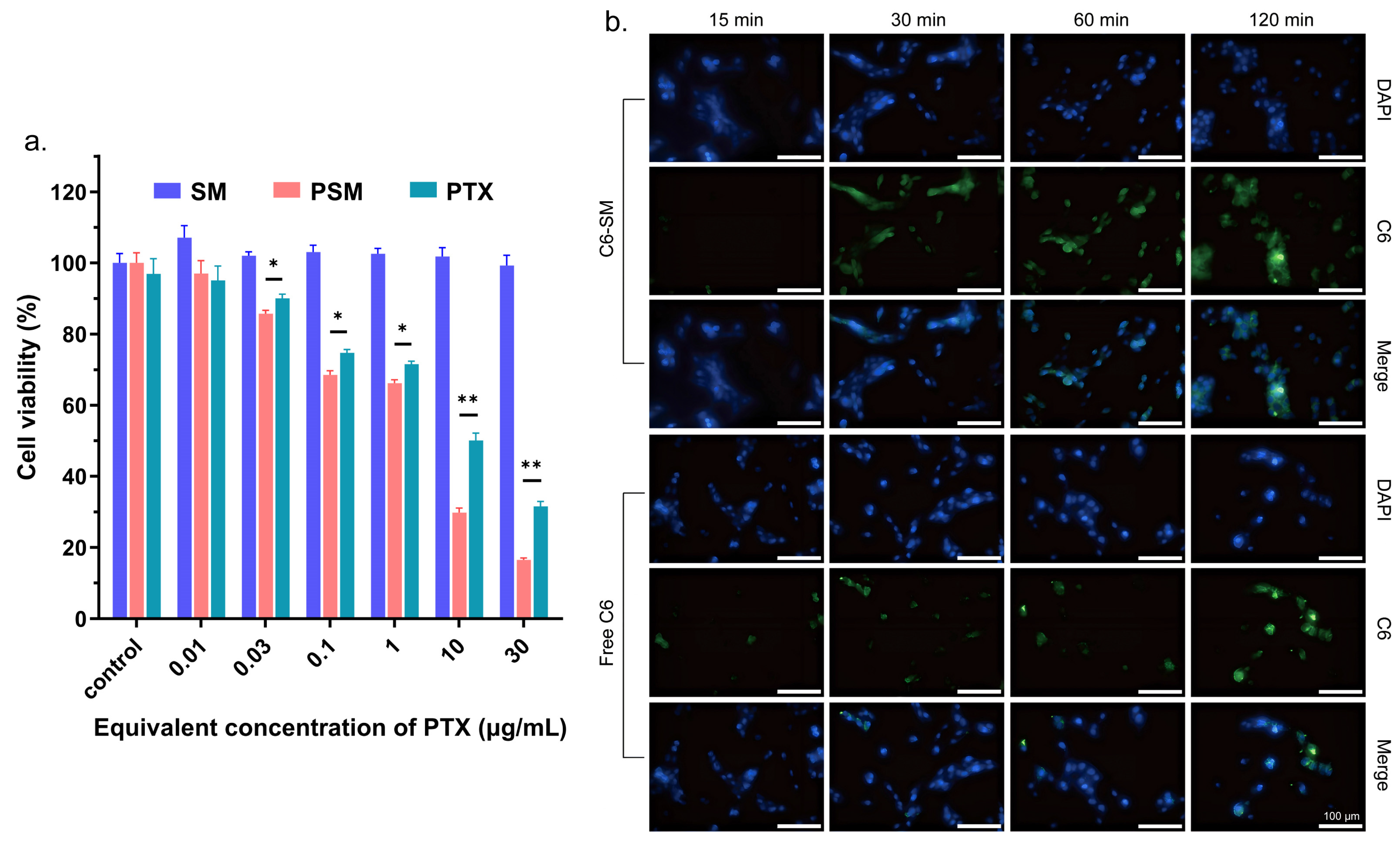
In vitro cytotoxicity: Firstly, the in vitro cytotoxicity of blank SM was evaluated by the CCK-8 method. C6 cells were inoculated at a density of 5 × 103 cells/well and incubated in 96-well plates for 24 h. Subsequently, the medium was replaced with SM equivalent to PTX concentrations of 30, 10, 1, 0.1, 0.03, and 0.01 μg/mL for 24 h. Next, CCK-8 was added and incubated for 2 h. The OD values of the solutions were recorded at 450 nm using an enzyme marker (BioTek, Dallas, TX, USA). Each group consisted of five parallel samples and cell viability was calculated as follows: Cell viability (%) = [(ODsample − ODblank)/(ODcontrol − ODblank)] × 100%. For the anti-cell proliferation assay of PSMs, the CCK-8 method was also used for evaluation. C6 cells were inoculated in 96-well plates at a density of 6 × 103 cells/well. After incubation for 24 h, the medium was replaced with PTX and PSMs equivalent to PTX concentrations of 30, 10, 1, 0.1, 0.03, and 0.01 μg/mL for 24 h and analysed as described above.
Cellular uptake: The micelles Cou-SMs (micelles loaded with Cou-6) were prepared by the same method using Cou-6 instead of PTX, and the particle size and potential were measured to indicate micelle formation. Logarithmic growth-phase C6 cells were inoculated into 12-well plates at a density of 1.5 × 105 cells/mL and cultured for 24 h. The old culture medium was aspirated, and the cell surface was washed with cold PBS buffer, and then 1 mL of serum-free culture medium diluted with 1 μg/mL of free Cou-6, Cou-SM was added to each group of cells, and the cells were incubated for 15, 30, 60, and 120 min, respectively. The medium was removed and discarded, and the cells were washed three times with cold PBS and then fixed with 4% paraformaldehyde for 15 min. We added 0.1% Triton X-100 and allowed the cells to permeabilize for 10 min. The nuclei were subsequently stained by adding DAPI. The fluorescence images of the cells were analysed using a cell imaging microplate detector (Cytation5, BioTek, Dallas, TX, USA), and light was avoided during the experiments.
2.4. Examination of Brain Targeting Efficiency
The construction of a mouse model bearing C6Luc glioma in situ: 5–8-week female BALB/c mice were taken and acclimatised for growth for one week in our laboratory. The mice were anaesthetised with 0.5 mL of Aphrodite for about 5 min and fixed on a mouse brain stereotaxic apparatus and the skin was incised through a midline sagittal incision to expose the skull. The surface tissue of the skull was then destroyed with 5% hydrogen peroxide to expose the skull. A single hole was drilled 1.8 mm to the right of the sagittal suture, approximately 3.0 mm deep. In total, 4 μL of logarithmic growth-phase C6Luc cells (~2 × 105 cells) were stereotactically inoculated into the target location at a position 4.0 mm below and 1.0 mm above the skull. Scalp incisions were closed by bone wax closure and surgical wounds were sterilised and sutured.
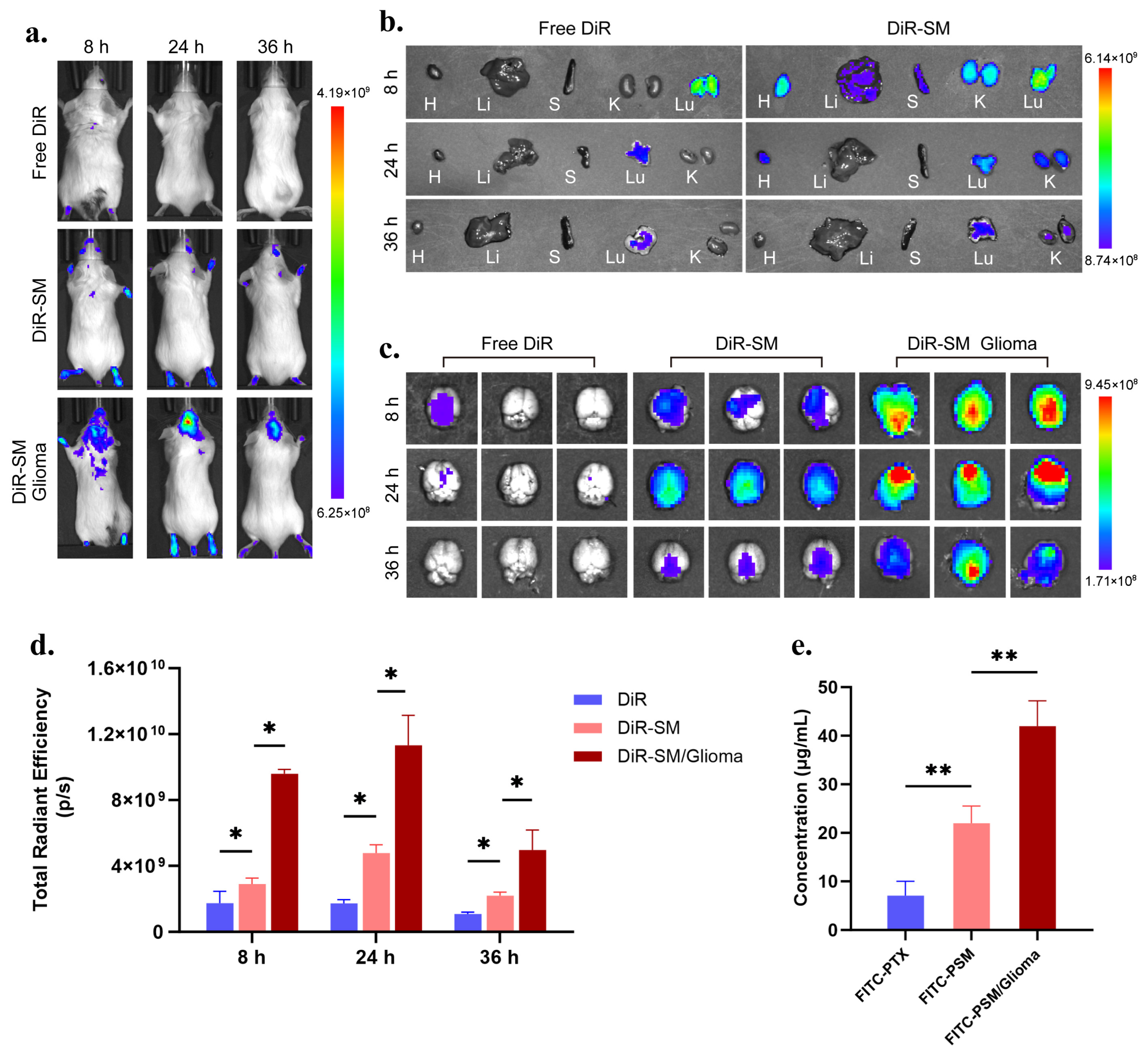
Brain targeting efficiency and in vivo distribution experiments: A DiR fluorescent probe was used instead of PTX, and micellar DSM (micelles loaded with DiR) was prepared by the same method. In total, 200 μL (0.5 mg/mL) of free DiR and DSM were injected into the tail vein of healthy mice and tumour-bearing mice, respectively, and the in vivo and organ fluorescence images were obtained from mice by the IVIS in vivo imaging system (Caliper Life Sciences Inc., Palo Alto, CA, USA) at 8, 24, and 36 h (Ex = 748 nm, Em = 780 nm). The images were processed using Living image 4.4 software, n = 3. Instead of PTX, FITC-PTX was used and micellar FITC-PSM was prepared by the same method. In total, 200 μL (1 mg/mL) of free FITC-PTX and FITC-PSM were injected into the tail vein of healthy and tumour-bearing mice, respectively, and the brain tissues were taken out and placed in methanol at 24 h. The brain tissues were processed by a grinder (Servicebio, SWE-3D, Wuhan, CHN). Tissue homogenates were prepared and the fluorescence intensity (Ex = 488 nm, Em = 525 nm) was measured using a multifunctional enzyme marker (BioTek., Dallas, TX, USA), and the drug concentration was obtained by banding it into the prepared standard curve, n = 3.
2.5. Examination of Anti-Tumour Activity and Safety in Mice In Vivo
Inhibition experiments on glioma in situ: Seven days after the inoculation of C6Luc cells, mice were randomly divided into a saline group, a positive Taxol group, a PSM group and a control group without tumour inoculation. Each group was injected with the corresponding drug saline solution at a PTX dose of 7.5 mg/kg every 3 days by tail vein injection, and 0.2 mL of PBS-diluted Luc luciferin substrate (15 mg/mL) was injected intraperitoneally on days 7, 10, 13, 16 and 19, and the tumour size was determined by the IVIS in vivo imaging system (Caliper Life Sciences Inc., Palo Alto, CA, USA) after 15 min. Bioluminescent signals were detected to determine tumour size, and the body weight of each group of mice was monitored once after each imaging. At the end of the treatment, the brain tissues were removed and photographed for observation of the tumours, which were then preserved in a universal tissue fixative, and paraffin sections were prepared and subjected to the TUNEL assay to check the apoptosis of the tumour cells.
In vivo safety evaluation: At the end of the last dose of the drug in mice, blood was taken from the eyeballs and serum samples were collected to determine the levels of ALT, AST, BUN, and CRE in order to assess the drug toxicity in mice. Mouse heart, liver, spleen, lung and kidney tissues were removed after the last drug administration and fixed in universal tissue fixative, followed by the preparation of paraffin sections for HE staining to observe the pathological changes and metastasis of tumour foci.
2.6. Statistical Analysis
The data are expressed as mean ± standard deviation (SD). Significant differences between two groups were tested by Student’s t-test. A one-way analysis of variance (ANOVA) and Bonferroni post hoc test were used for comparison between groups. Data graphs were prepared and analysed using Prism 10.1.2 software. * p < 0.05, ** p < 0.01, *** p < 0.001, ns for p > 0.05.
4. Discussion
PTX, as a broad-spectrum antitumour agent, has not been approved for the clinical treatment of gliomas, although it has demonstrated significant efficacy in the treatment of a wide range of malignant tumours, mainly due to its difficulty in achieving effective therapeutic concentrations in brain tumour lesions [
22]. Although some studies have reported that Taxol can detect PTX accumulation in the focal tissues of glioma patients, the levels in normal brain tissues are below the limit of detection (thanks to the permeability of the BBTB and the micellar properties of Taxol) [
23]. Due to its serious adverse effects, Taxol has been gradually replaced by injectable paclitaxel liposomes and Abraxane
®. However, the larger particle size of the latter two rather limits their BBTB penetration ability. Most glioma studies have focused on breaching the BBB, ignoring the fact that the BBTB is the main obstacle to glioma treatment [
18]. Existing targeting strategies (e.g., ligand modification, magnetic targeting, ultrasound-assisted or transnasal drug delivery) [
24,
25] are mainly aimed at the penetration of the intact BBB, and these approaches, although suitable for the treatment of neurodegenerative diseases, are not optimal for gliomas. Although these techniques can facilitate drug entry into the brain and navigation to the tumour site, the resulting distribution of the drug throughout the brain may cause damage to normal brain tissue, and the complex preparation process hinders clinical translation.
In this study, we systematically compared the penetration ability of nanomicelles in healthy mice (BBB model) and tumour-bearing mice (the BBTB model) using DiR and FITC-PTX as tracers. The results showed that nanomicelles exhibited stronger brain targeting in hormonal mice compared to free tracers. Notably, the nanocolloids showed whole-brain distribution in healthy mice, whereas they specifically accumulated in tumour foci in model mice, confirming that small-particle-size nanocolloids can penetrate the BBTB by passive targeting and enrich at tumour sites with the help of the EPR effect.
By establishing a C6Luc in situ glioma model, we comparatively assessed the anti-tumour effects of PSM versus Taxol. The experimental data showed that the PSM treatment group exhibited more significant tumour growth inhibition and stronger pro-apoptotic effects than the Taxol group. Meanwhile, the effects of PSMs on body weight, liver and kidney functions, and major organs of mice were significantly smaller than those of Taxol. Although Taxol also has a micellar structure, which theoretically should have the ability to penetrate the BBTB, its antitumour effect was weaker than that of PSMs in this study. This phenomenon may be related to Taxol-induced systemic toxicity, which may weaken the body’s natural defences against malignancy.



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}