

Comprehensive Investigation of Polymorphic Stability and Phase Transformation Kinetics in Tegoprazan


 ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Computational Methods for Conformational and Energetic Analysis
2.3. NMR Measurements
2.4. Solubility Measurement
2.5. High-Resolution PXRD (HR-PXRD) and Time-Dependent HR-PXRD Study of SMPT
2.6. DSC Analysis of SMPT
3. Results and Discussion
3.1. Summary of Key Findings
3.1.1. Polymorph Identification and Thermodynamic Stability
- Three solid-state forms of TPZ were studied: Polymorph A, Polymorph B, and the amorphous form.
- Polymorph A was consistently identified as the thermodynamically stable form under all tested conditions and solvents.
- Polymorph B was observed only transiently in aprotic solvents such as acetone and MEK, indicating its metastable nature.
- The amorphous form exhibited higher solubility but converted readily to Polymorph A, especially under accelerated storage or slurry conditions.
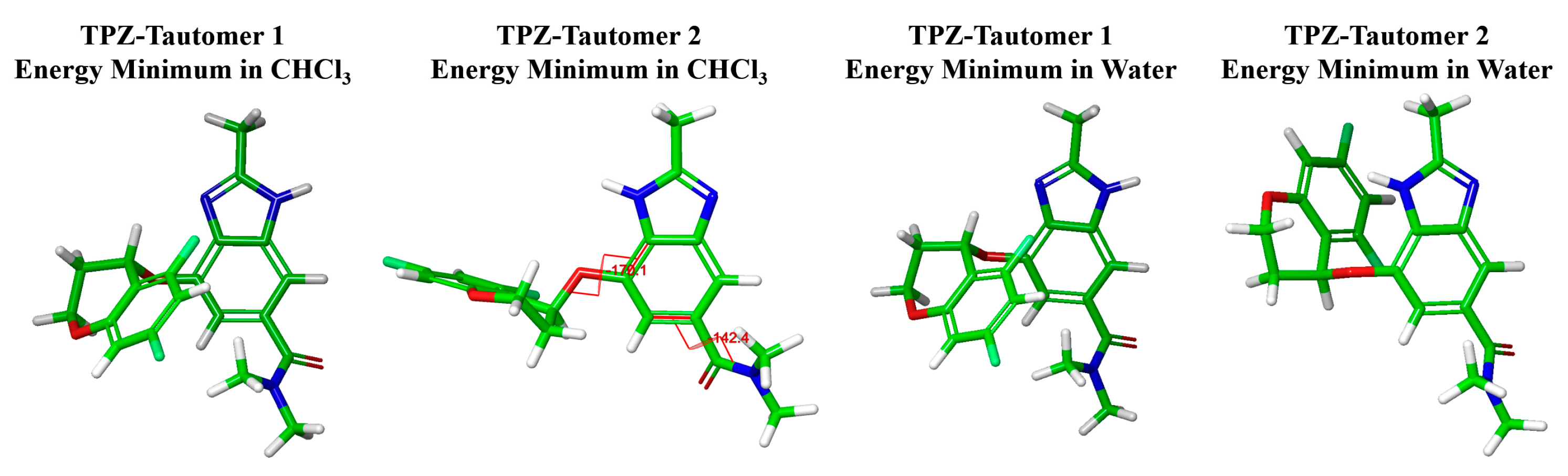
- Solution-phase conformational preferences and tautomerism
- Conformational energy landscape mapping combined with DFT-D calculations showed that the most stable solution-phase conformers strongly resembled the structures observed in Polymorph A.
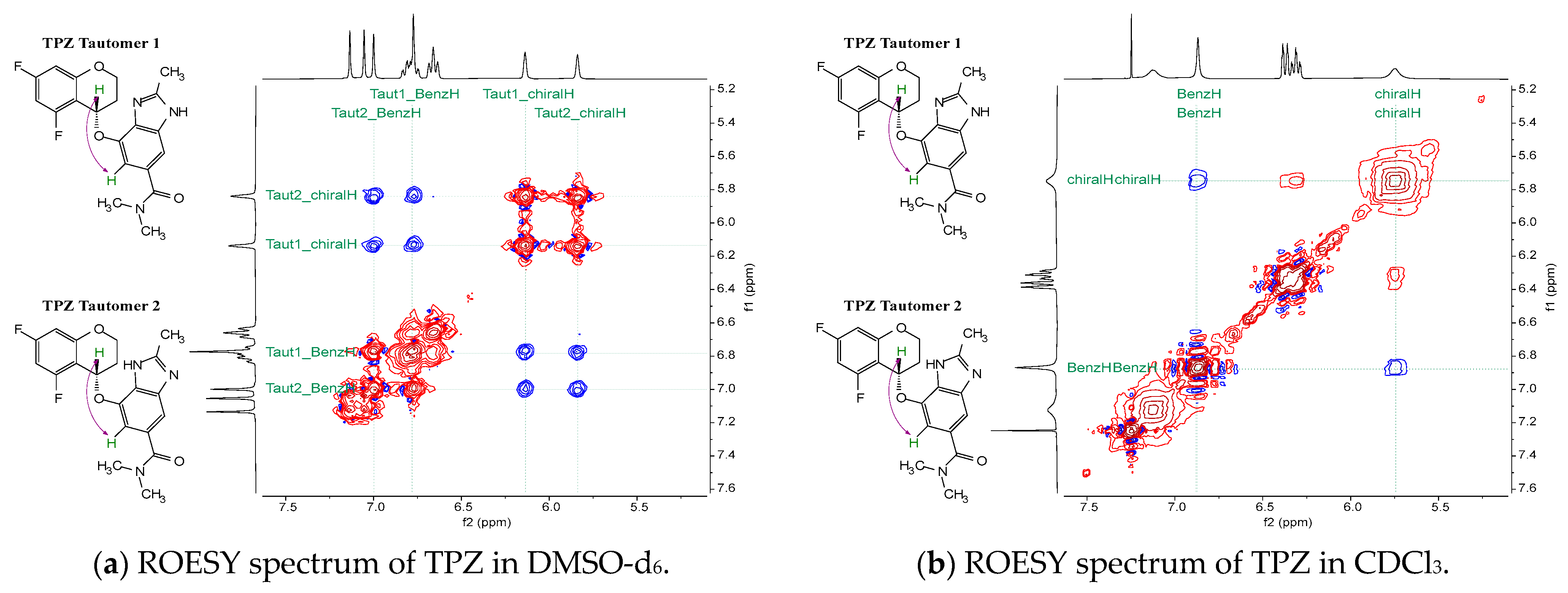
- TPZ exists as two tautomeric forms in solution. Tautomer 2 predominates in protic solvents and adopts a conformation aligned with Polymorph A.
- ROESY NMR spectra confirmed solvent-dependent NOE patterns, reflecting tautomer-specific conformational arrangements.
3.1.2. Hydrogen Bonding and Crystal Packing
- DFT-D single-point energy calculations of hydrogen-bonded dimers extracted from crystal structures demonstrated that dimers in Polymorph A are energetically more favorable overall.
- Although one specific dimer motif in Polymorph B showed the lowest individual energy, it did not appear to drive stable crystallization under experimental conditions.
3.1.3. Solubility and SMPT
- Solubility measurements revealed that both Polymorph B and the amorphous form gradually transformed into Polymorph A in all solvents tested (acetone, methanol, and water), confirming thermodynamic convergence.
- Time-dependent PXRD monitoring of suspensions captured the progression of SMPT from B or the amorphous form to Polymorph A. Importantly, no unknown intermediate phases or alternative crystal forms exhibiting greater stability than Polymorph A were detected under any of the tested conditions.
3.1.4. Kinetic Modeling Using the KJMA Equation
- Transformation kinetics were successfully modeled using the KJMA equation, allowing the extraction of the rate constant (k) and Avrami exponent (n).
- Solvent-dependent differences were observed:
- ○
- Methanol: fastest transformation (k = 0.22, n = 11.3 from amorphous).
- ○
- Acetone: moderate kinetics with intermediate B phase (n = 5.5–8.4).
- ○
- Water: slow transformation (n = 6.3–11.5), requiring DSC-based estimation.
3.1.5. Mechanistic Implications and Polymorph Selection
- Solvent polarity and hydrogen-bond donor ability influenced both tautomer distribution and conformational bias, which in turn affected the polymorph outcome.
- In methanol, strong dual hydrogen bonding suppressed the formation of Polymorph B, leading directly to Polymorph A.
- In acetone, weaker solvent interactions allowed the transient stabilization of Polymorph B, which then transformed to A via a two-step pathway.
- These findings support a mechanism in which solution-phase conformers and solvent interactions collectively direct polymorph selection.
- The absence of detectable Polymorph B in protic solvents may reflect a “disappearing polymorph” scenario, a critical concern in pharmaceutical solid-state design.
3.2. Computational Investigation of Molecular Interactions and Stability in Solution
3.2.1. Computational Analysis of Solvent-Phase Conformational Preferences
3.2.2. Correlation Between Solution Conformers and NOE Observations
3.2.3. Hydrogen Bonding Mechanisms in Crystallization Guided by Solution Conformers
3.3. Solubility Measurements and Thermodynamic Stability of Polymorphs
3.4. Phase Transformation Monitoring Under Suspension Conditions by HR-PXRD
3.4.1. Time-Dependent HR-PXRD Analysis of Polymorph B Suspension
3.4.2. Time-Dependent HR-PXRD Analysis of Amorphous Suspension
3.4.3. Solvent-Directed Tautomerization and Its Influence on Polymorph Selection
3.5. Broader Context and Future Perspectives
3.5.1. Structural Comparison with Other P-CABs and Mechanistic Implications
- Solvent-mediated phase transitions (SMPTs) with transient intermediate states.
- Disappearing polymorphs, particularly Polymorph B, under protic solvent conditions.
- Broader conformational landscapes, increasing the unpredictability of crystallization.
3.5.2. Study Limitations and Future Directions
Study Limitations
- The conformational and hydrogen-bonding analyses were conducted only for selected solvents (chloroform and water), while methanol and acetone—despite their demonstrated experimental significance—were not explicitly included in DFT-D modeling. This limitation arose due to restrictions in the solvent models available within the force field and PCM settings of the software used (OPLS4 and Jaguar), which do not provide comprehensive parameterization for all solvents at the quantum mechanical level.
- Hydrogen bonding was analyzed at the dimer level without incorporating periodic boundary conditions or full lattice energy calculations, limiting direct comparisons of overall crystal packing stability. This was primarily due to computational resource limitations, as periodic DFT-D calculations for complex organic crystals—particularly with a large asymmetric unit (Z′ = 2)—require substantial memory and CPU time that exceeded the available system capabilities.
- The tautomeric equilibrium was inferred primarily from NOE-based NMR and supported by solution-phase modeling, but the quantitative measurement of tautomer populations in various solvents was not performed.
- Comparative analyses with other P-CABs were limited to structural features; the polymorphic behaviors of vonoprazan, revaprazan, and soraprazan were not studied experimentally under analogous crystallization conditions.
Future Directions
- Extend DFT-D conformational and hydrogen-bonding analyses to additional solvents (e.g., methanol, acetone) as software capabilities evolve to quantitatively link solvent effects with polymorph selectivity.
- Perform full periodic DFT optimizations and lattice energy calculations for each polymorph, once sufficient computational resources are available, to rigorously assess thermodynamic stability and crystal packing forces. These high-level calculations will also enable comparative evaluation against current lower-cost approaches—such as dimer-based DFT-D and force-field-based methods—to determine whether such approximations can reliably capture relative polymorph stability. Establishing this correlation would validate the continued use of computationally efficient methods in early-stage polymorph screening, especially for compounds with large unit cells or limited resources.
- Expand the comparative study to other P-CABs (e.g., vonoprazan, revaprazan, soraprazan) by applying the same analytical workflow. Although these compounds do not exhibit tautomerization, parallel studies could reveal whether other subtle conformational or hydrogen-bonding effects influence polymorph selection and transformation kinetics in this drug class.
- More broadly, the role of tautomerism in polymorphic diversity warrants systematic investigation across tautomeric APIs, with TPZ serving as a mechanistic model for tautomer-driven crystallization.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| API | Active Pharmaceutical Ingredient; |
| CI | Confidence Interval; |
| CSP | Crystal Structure Prediction; |
| DFT-D | Density Functional Theory with Dispersion Correction; |
| DSC | Differential Scanning Calorimetry; |
| HPLC | High-Performance Liquid Chromatography; |
| HR-PXRD | High-Resolution PXRD; |
| ICH Q2(R1) | International Council for Harmonisation Guideline Q2(R1): Validation of Analytical Procedures; |
| KJMA | Kolmogorov–Johnson–Mehl–Avrami; |
| LOD | Limit of Detection; |
| LOQ | Limit of Quantitation; |
| NMR | Nuclear Magnetic Resonance; |
| NOE | Nuclear Overhauser Effect; |
| OLS | Ordinary Least Squares; |
| OPLS4 | Optimized Potentials for Liquid Simulations 4; |
| PCM | Polarizable Continuum Model; |
| PI | Prediction Interval; |
| P-CAB | Potassium-Competitive Acid Blocker; |
| PXRD | Powder X-ray Diffraction; |
| ROESY | Rotating-Frame Overhauser effect spectroscopy; |
| Rwp | Weighted Profile R-factor; |
| Rexp | Expected R-factor; |
| SDPD | Structure Determination by Powder Diffractometry; |
| SMPT | Solvent-Mediated Phase Transformation; |
| TGA | Thermogravimetric Analysis; |
| TPZ | Tegoprazan; |
| Z′ | Number of independent molecules in asymmetric unit (crystallography term). |
References
- Dunitz, J.D.; Bernstein, J. Disappearing Polymorphs. Acc. Chem. Res. 1995, 28, 193–200. [Google Scholar] [CrossRef]
- Rubin-Preminger, J.M.; Bernstein, J. 3-Aminobenzenesulfonic Acid: A Disappearing Polymorph. Cryst. Growth Des. 2005, 5, 1343–1349. [Google Scholar] [CrossRef]
- Blagden, N.; Davey, R.J.; Rowe, R.; Roberts, R. Disappearing Polymorphs and the Role of Reaction By-Products: The Case of Sulphathiazole. Int. J. Pharm. 1998, 172, 169–177. [Google Scholar] [CrossRef]
- Malec, L.M.; Gryl, M.; Oszajca, M.T.; Brela, M.Z.; Stadnicka, K.M. Chasing the Co-Crystal Disappearing Polymorph with Ab Initio Methods. Cryst. Growth Des. 2021, 21, 6902–6912. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals, 2nd ed.; Oxford University Press: Oxford, UK, 2020; ISBN 9780199655441. [Google Scholar]
- Bučar, D.; Lancaster, R.W.; Bernstein, J. Verschwundene Polymorphe: Eine Neubetrachtung. Angew. Chem. 2015, 127, 7076–7098. [Google Scholar] [CrossRef]
- Sacchi, P.; Wright, S.E.; Neoptolemou, P.; Lampronti, G.I.; Rajagopalan, A.K.; Kras, W.; Evans, C.L.; Hodgkinson, P.; Cruz-Cabeza, A.J. Crystal Size, Shape, and Conformational Changes Drive Both the Disappearance and Reappearance of Ritonavir Polymorphs in the Mill. Proc. Natl. Acad. Sci. USA 2024, 121, e2319127121. [Google Scholar] [CrossRef] [PubMed]
- FDA Novartis Issues Voluntary US Nationwide Recall of Two Lots of Sandimmune® Oral Solution (Cyclosporine Oral Solution, USP), 100 Mg/ML Due to Crystallization. Available online: https://www.novartis.com/report (accessed on 21 March 2025).
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Hilfiker, R. Polymorphism: In the Pharmaceutical Industry, 1st ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2006; ISBN 3527311467. [Google Scholar] [CrossRef]
- Price, S.L. Predicting Crystal Structures of Organic Compounds. Chem. Soc. Rev. 2014, 43, 2098–2111. [Google Scholar] [CrossRef] [PubMed]
- Mattei, A.; Hong, R.S.; Dietrich, H.; Firaha, D.; Helfferich, J.; Liu, Y.M.; Sasikumar, K.; Abraham, N.S.; Bhardwaj, R.M.; Neumann, M.A.; et al. Efficient Crystal Structure Prediction for Structurally Related Molecules with Accurate and Transferable Tailor-Made Force Fields. J. Chem. Theory Comput. 2022, 18, 5725–5738. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, M.; Hoja, J.; Aerts, L.; Quéré, L.; van de Streek, J.; Neumann, M.A.; Tkatchenko, A. Computational Polymorph Screening Reveals Late-Appearing and Poorly-Soluble Form of Rotigotine. Commun. Chem. 2019, 2, 70. [Google Scholar] [CrossRef]
- Nyman, J.; Day, G.M. Static and Lattice Vibrational Energy Differences between Polymorphs. CrystEngComm 2015, 17, 5154–5165. [Google Scholar] [CrossRef]
- Price, S.L. Is Zeroth Order Crystal Structure Prediction (CSP_0) Coming to Maturity? What Should We Aim for in an Ideal Crystal Structure Prediction Code? Faraday Discuss. 2018, 211, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Take, Y. Tegoprazan, a Novel Potassium-Competitive Acid Blocker to Control Gastric Acid Secretion and Motility. J. Pharmacol. Exp. Ther. 2018, 364, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Lee, K.H.; Kim, S.J.; Kim, S.J.; Lee, S.J.; Park, C.H.; Kim, B.T.; Song, G.S.; Moon, B.S.; Ryu, S.Y. Effects of Tegoprazan, a Novel Potassium-Competitive Acid Blocker, on Rat Models of Gastric Acid-Related Disease. J. Pharmacol. Exp. Ther. 2019, 369, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, E.S.; Lee, Y.; Lee, W.; Kweon, H.; Lee, S.A.; Do Choi, K.; Ko, D.H.; Heo, S.P. Crystalline Form of a Benzimidazole Derivative and a Preparation Method Thereof. U.S. Patent 009908870B2, 6 March 2018. [Google Scholar]
- Cao, M.; Chen, D.; Zhang, Z.; Chen, Y.; Jin, F.; Yang, S.; Zheng, B.; Gao, Q. Tegoprazan Crystal Form B and Preparation Method Therefor. Patent WO2024045255A1, 7 March 2024. [Google Scholar]
- Ryu, S.; Lee, J.H.; Kim, J.; Yamaguchi, T. Structure Determination of Tegoprazan((S)-4-((5,7-Difluorochroman-4-Yl)Oxy)-N, N,2-Trimethyl-1H-Benzo[d]Imidazole-6-Formamide) Polymorphs A and B by Laboratory X-Ray Powder Diffraction. Molecules 2025, 30, 1538. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Spong, B.; Price, C.P.; Jayasankar, A.; Matzger, A.J.; Rodríguez-Hornedo, N. General Principles of Pharmaceutical Solid Polymorphism: A Supramolecular Perspective. Adv. Drug Deliv. Rev. 2004, 56, 241–274. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Grant, D.J.W. Influence of Crystal Structure on the Tableting Properties of Sulfamerazine Polymorphs. Pharm. Res. 2001, 18, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Griesser, U.J. The Importance of Solvates. In Polymorphism: In the Pharmaceutical Industry; Hilfiker, R., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2006; pp. 211–233. ISBN 3527311467/9783527607884. [Google Scholar]
- Kitamura, M. Controlling Factors and Mechanism of Polymorphic Crystallization. Cryst. Growth Des. 2004, 4, 1153–1159. [Google Scholar] [CrossRef]
- Khawam, A.; Flanagan, D.R. Solid-State Kinetic Models: Basics and Mathematical Fundamentals. J. Phys. Chem. B 2006, 110, 17315–17328. [Google Scholar] [CrossRef] [PubMed]
- Andrés, A.; Rosés, M.; Ràfols, C.; Bosch, E.; Espinosa, S.; Segarra, V.; Huerta, J.M. Setup and Validation of Shake-Flask Procedures for the Determination of Partition Coefficients (LogD) from Low Drug Amounts. Eur. J. Pharm. Sci. 2015, 76, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Doebelin, N.; Kleeberg, R. Profex: A Graphical User Interface for the Rietveld Refinement Program BGMN. J. Appl. Crystallogr. 2015, 48, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.P.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures. J. Chem. Theory Comput. 2018, 14, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Heimsaat, M. Intermolecular Interactions in Molecular Organic Crystals upon Relaxation of Lattice Parameters. Crystals 2019, 9, 665. [Google Scholar] [CrossRef]
- Wappett, D.A.; Goerigk, L. Benchmarking Density Functional Theory Methods for Metalloenzyme Reactions: The Introduction of the MME55 Set. J. Chem. Theory Comput. 2023, 19, 8365–8383. [Google Scholar] [CrossRef] [PubMed]
- Marchese Robinson, R.L.; Geatches, D.; Morris, C.; MacKenzie, R.; Maloney, A.G.P.; Roberts, K.J.; Moldovan, A.; Chow, E.; Pencheva, K.; Vatvani, D.R.M. Evaluation of Force-Field Calculations of Lattice Energies on a Large Public Dataset, Assessment of Pharmaceutical Relevance, and Comparison to Density Functional Theory. J. Chem. Inf. Model. 2019, 59, 4778–4792. [Google Scholar] [CrossRef] [PubMed]
- Dorigo, A.E.; Pratt, D.W.; HouK, K.N. Origin of Methyl Conformational Preferences and Rotational Barriers in the Ground States, Excited Triplet States, Radical Cations, and Radical Anions of Molecules Having CH3-C:X Functionalities. J. Am. Chem. Soc. 1987, 109, 6591–6600. [Google Scholar] [CrossRef]
- Nam, S.; Cho, E.; Burke, K.; Sim, E. Explaining and Fixing DFT Failures for Torsional Barriers. J. Phys. Chem. Lett. 2021, 12, 2796–2804. [Google Scholar] [CrossRef] [PubMed]
- Stylianakis, I.; Zervos, N.; Lii, J.H.; Pantazis, D.A.; Kolocouris, A. Conformational Energies of Reference Organic Molecules: Benchmarking of Common Efficient Computational Methods against Coupled Cluster Theory. J. Comput.-Aided Mol. Des. 2023, 37, 607–656. [Google Scholar] [CrossRef] [PubMed]
- Kleinpeter, E. NMR Spectroscopic Study of Tautomerism in Solution and in the Solid State. In Tautomerism: Methods and Theories; Antonov, L., Ed.; Wiley: Hoboken, NJ, USA, 2013; pp. 103–143. ISBN 9783527658824. [Google Scholar]
- García, M.Á.; Claramunt, R.M.; Solčan, T.; Milata, V.; Alkorta, I.; Elguero, J. 13C and 15N NMR Spectra of Aminobenzimidazoles in Solution and in the Solid State. Magn. Reson. Chem. 2009, 47, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Řezáč, J.; Šimová, L.; Hobza, P. CCSD[T] Describes Noncovalent Interactions Better than the CCSD(T), CCSD(TQ), and CCSDT Methods. J. Chem. Theory Comput. 2013, 9, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Ladóczki, B.; Gyevi-Nagy, L.; Nagy, P.R.; Kállay, M. Enabling Accurate and Large-Scale Explicitly Correlated CCSD(T) Computations via a Reduced-Cost and Parallel Implementation. J. Chem. Theory Comput. 2025, 21, 2432–2447. [Google Scholar] [CrossRef] [PubMed]
- Young, R.A. The Rietveld Method; Oxford University Press: Oxford, UK, 1995; ISBN 9780198559122. [Google Scholar]
- David, W.I.F.; Shankland, K.; McCusker, L.B.; Baerlocher, C. Structure Determination from Powder Diffraction Data; Oxford University Press: Oxford, UK, 2002; ISBN 9780198500919. [Google Scholar]
- Toby, B.H. R Factors in Rietveld Analysis: How Good Is Good Enough? Powder Diffr. 2006, 21, 67–70. [Google Scholar] [CrossRef]
- Sangwal, K. Phase Transformation and Isothermal Crystallization Kinetics. In Nucleation and Crystal Growth: Metastability of Solutions and Melts; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 145–188. [Google Scholar]
- Avrami, M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of Phase Change. II Transformation-Time Relations for Random Distribution of Nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, Phase Change, and Microstructure Kinetics of Phase Change. III. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Alexander, L.; Klug, H.P. Basic Aspects Of X-Ray Absorption In Quantitative Diffraction Analysis of Powder Mixtures. Anal. Chem. 1948, 20, 886–889. [Google Scholar] [CrossRef]
- Lappalainen, M.; Karppinen, M. Techniques of Differential Scanning Calorimetry for Quantification of Low Contents of Amorphous Phases. J. Therm. Anal. Calorim. 2010, 102, 171–180. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen Bridges in Crystal Engineering: Interactions without Borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Max, J.J.; Chapados, C. Infrared Spectroscopy of Acetone-Methanol Liquid Mixtures: Hydrogen Bond Network. J. Chem. Phys. 2005, 122, 014504. [Google Scholar] [CrossRef] [PubMed]
- Idrissi, A.; Polok, K.; Gadomski, W.; Vyalov, I.; Agapov, A.; Kiselev, M.; Barj, M.; Jedlovszky, P. Detailed Insight into the Hydrogen Bonding Interactions in Acetone-Methanol Mixtures. A Molecular Dynamics Simulation and Voronoi Polyhedra Analysis Study. Phys. Chem. Chem. Phys. 2012, 14, 5979–5987. [Google Scholar] [CrossRef] [PubMed]
- Dematos, L.L.; Williams, A.C.; Booth, S.W.; Petts, C.R.; Taylor, D.J.; Blagden, N. Solvent Influences on Metastable Polymorph Lifetimes: Real-Time Interconversions Using Energy Dispersive X-Ray Diffractometry. J. Pharm. Sci. 2007, 96, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Bobrovs, R.; Seton, L.; Actiņš, A. Solvent-Mediated Phase Transformation between Two Tegafur Polymorphs in Several Solvents. CrystEngComm 2014, 16, 10581–10591. [Google Scholar] [CrossRef]
- Gu, C.-H.; Young, V.; Grant, D.J.W. Polymorph Screening: Influence of Solvents on the Rate of Solvent-Mediated Polymorphic Transformation. J. Pharm. Sci. 2001, 90, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Simon, W.A.; Herrmann, M.; Klein, T.; Shin, J.M.; Huber, R.; Senn-Bilfinger, J.; Postius, S. Soraprazan: Setting New Standards in Inhibition of Gastric Acid Secretion. J. Pharmacol. Exp. Ther. 2007, 321, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qin, Z.; Meng, Y.; Cui, J.; Qian, L. Vonoprazan Fumarate Monocrystalline and Its Production and Use. Patent CN106478597A, 8 March 2017. [Google Scholar]
- Kuang, S.; Chen, N.; Wang, Y.; Yun, X. A Kind of Vonoprazan Fumarate Novel Crystal Forms and Preparation Method Thereof. Patent CN106478601A, 8 March 2017. [Google Scholar]
- Liu, W.; Wang, G.; Hou, Q.; Meng, H. Crystalline Polymorph of Revaprazan Hydrochloride and Its Preparation Method. Patent JP2017534635A, 24 November 2017. [Google Scholar]
- Liu, W.; Wang, G.; Hou, Q.; Meng, H. Revaprazan Hydrochloride Polymorphs and Preparation Therefor. U.S. Patent 2017/0334877 A1, 23 November 2017. [Google Scholar]
- Lee, M.J.; Kim, J.Y.; Kim, P.; Lee, I.S.; Mswahili, M.E.; Jeong, Y.S.; Choi, G.J. Novel Cocrystals of Vonoprazan: Machine Learning-Assisted Discovery. Pharmaceutics 2022, 14, 429. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ozako, M.; Inukai, M.; Matsuyuki, Y.; Kitayama, S.; Kanai, C.; Nagai, C.; Gopalasingam, C.C.; Gerle, C.; Shigematsu, H.; et al. Deep Learning Driven de Novo Drug Design Based on Gastric Proton Pump Structures. Commun. Biol. 2023, 6, 956. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Morita, M.; Yamagishi, T.; Madapally, H.V.; Hayashida, K.; Khandelia, H.; Gerle, C.; Shigematsu, H.; Oshima, A.; Abe, K. Structural Basis for Binding of Potassium-Competitive Acid Blockers to the Gastric Proton Pump. J. Med. Chem. 2022, 65, 7843–7853. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Angle Type | Energy Minimum in CHCl3 | Energy Minimum in Water | Individual Conformers in Polymorphs A and B | |||||
|---|---|---|---|---|---|---|---|---|
| Dihedral angle (°) | TPZ-Tautomer | TPZ_Polymorph | ||||||
| Torsion type | 1 | 2 | 1 | 2 | A1 | A2 | B1 | B2 |
| Chromane side | −72.92 (287.08) | −170.09 (189.91) | −73.56 (286.44) | 91.94 | 168.71 | 55.05 | −11.61 (348.39) | 3.40 |
| Amide side | −44.42 (315.58) | −142.43 (217.57) | −46.96 (313.04) | −45.94 (314.06) | −127.91 (232.09) | 114.63 | −138.60 (221.40) | 134.03 |
| Polymorph Type | TPZ_Polymorph A | TPZ_Polymorph B | ||
|---|---|---|---|---|
| Hydrogen-bonding type | 1 | 2 | 1 | 2 |
| ΔE (kcal/mol) | 2.49 | 3.37 | 0.00 | 12.44 |
| Solvent | Polymorph A (mg/mL) | Polymorph B (mg/mL) | Amorphous (mg/mL) |
|---|---|---|---|
| Acetone | 10 | 520 | 660 |
| Methanol | 257 | 552 | 580 |
| Water | 0.035 | 0.116 | 0.600 |
| Solvent | KJMA Equation | Rate Constant (k) | Avrami Exponent (n) |
|---|---|---|---|
| Acetone | Y = 1 − exp(−0.932t5.5) | 0.932 | 5.5 |
| Methanol | Y = 1 − exp(−12.76t1.24) | 12.76 | 1.24 |
| Water | Y = 1 − exp(−4.28 × 10−9t6.3) | 4.28 × 10−9 | 6.3 |
| Solvent | KJMA Equation | Rate Constant (k) | Avrami Exponent (n) |
|---|---|---|---|
| Acetone | Y = 1 − exp(−1.84 × 10−3t8.42) | 1.84 × 10−3 | 8.42 |
| Methanol | Y = 1 − exp(−0.22t11.3) | 0.22 | 11.3 |
| Water | Y = 1 − exp(−3.94 × 10−28t11.5) | 3.94 × 10−28 | 11.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Kim, K.H.; Ryu, S.A.; Kim, J.; Jung, K.; Kang, K.S.; Yamaguchi, T. Comprehensive Investigation of Polymorphic Stability and Phase Transformation Kinetics in Tegoprazan. Pharmaceutics 2025, 17, 928. https://doi.org/10.3390/pharmaceutics17070928
Lee JH, Kim KH, Ryu SA, Kim J, Jung K, Kang KS, Yamaguchi T. Comprehensive Investigation of Polymorphic Stability and Phase Transformation Kinetics in Tegoprazan. Pharmaceutics. 2025; 17(7):928. https://doi.org/10.3390/pharmaceutics17070928
Chicago/Turabian StyleLee, Joo Ho, Ki Hyun Kim, Se Ah Ryu, Jason Kim, Kiwon Jung, Ki Sung Kang, and Tokutaro Yamaguchi. 2025. "Comprehensive Investigation of Polymorphic Stability and Phase Transformation Kinetics in Tegoprazan" Pharmaceutics 17, no. 7: 928. https://doi.org/10.3390/pharmaceutics17070928
APA StyleLee, J. H., Kim, K. H., Ryu, S. A., Kim, J., Jung, K., Kang, K. S., & Yamaguchi, T. (2025). Comprehensive Investigation of Polymorphic Stability and Phase Transformation Kinetics in Tegoprazan. Pharmaceutics, 17(7), 928. https://doi.org/10.3390/pharmaceutics17070928