
Could Sodium-Glucose Co-Transporter-2 Inhibitors and Glucagon-like Peptide-1 Receptor Agonists Play a Role in Gout Treatment?


Abstract
1. Introduction
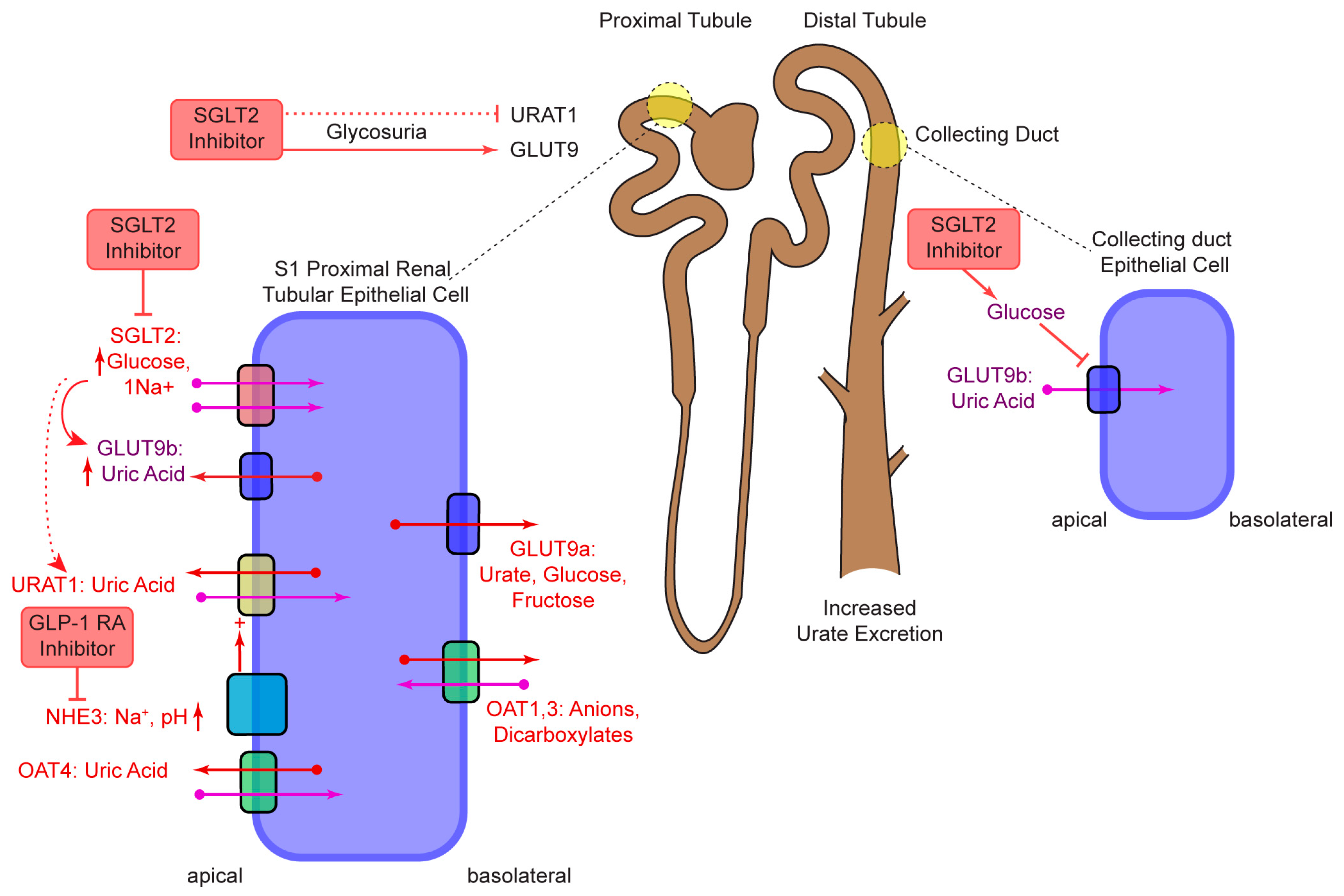
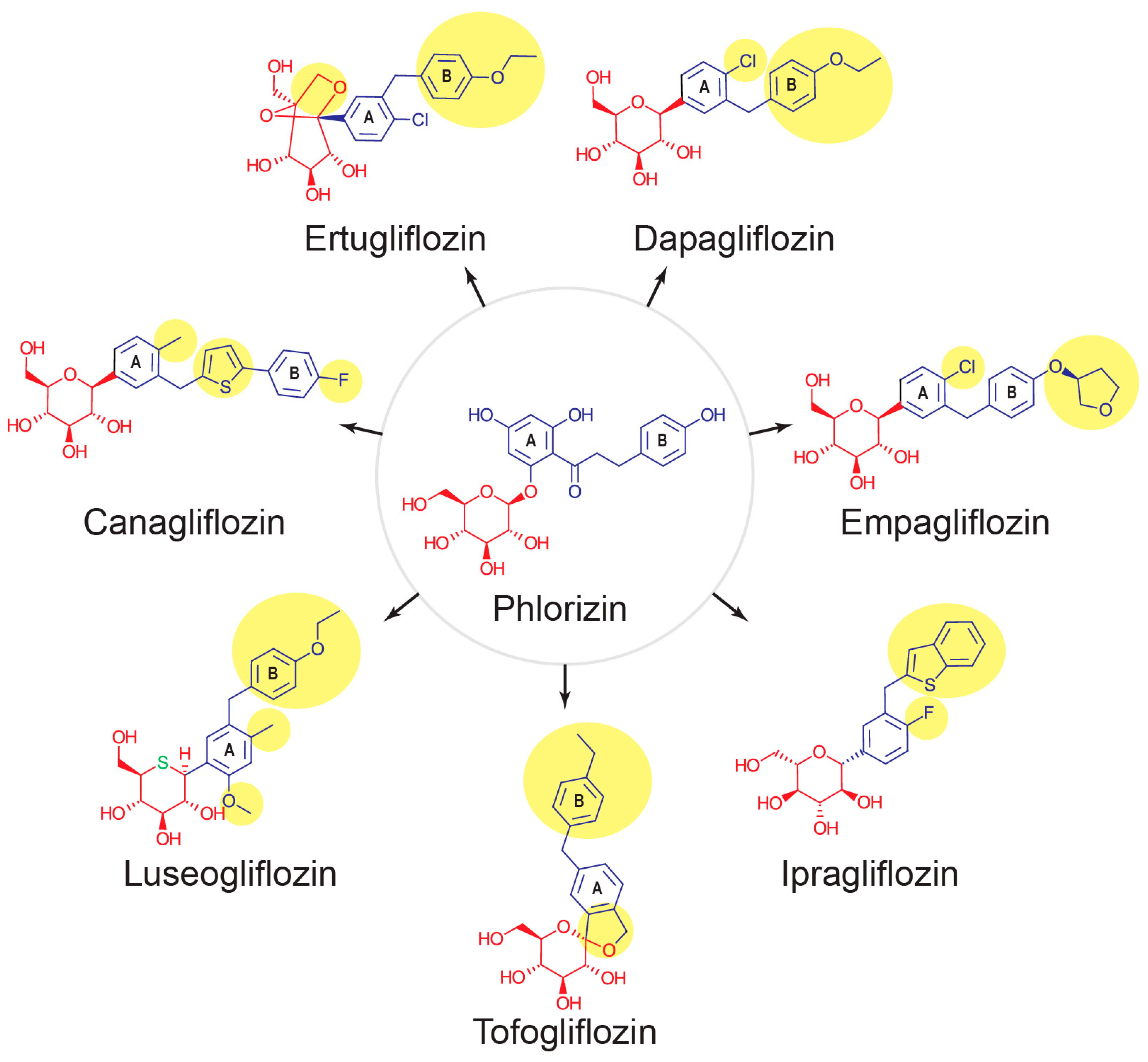
2. SGLT2
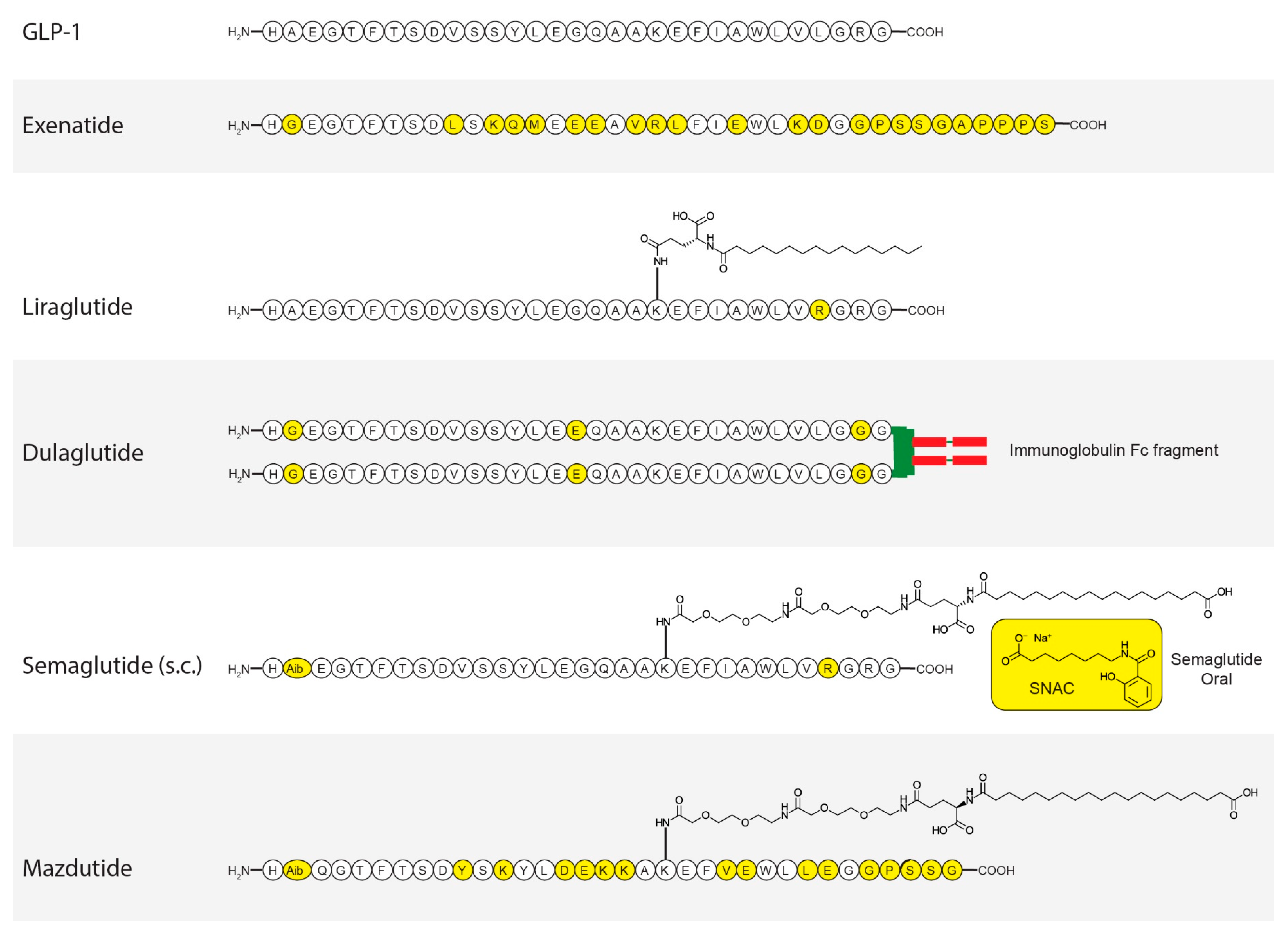
3. GLP-1
4. Approved SGLT2 Inhibitors, Serum Urate Lowering, and Their Possible Roles as Gout Treatments
4.1. Canagliflozin
4.2. Empagliflozin
4.3. Dapagliflozin
4.4. Luseogliflosin
4.5. Ipragliflozin
4.6. Tofogliflozin
4.7. Ertugliflozin
5. SGLT2 Inhibitors in the Pipeline
6. Approved GLP-1RAs, Serum Urate Lowering, and Their Possible Roles as Gout Treatments
6.1. Exenatide
6.2. Dulaglutide
6.3. Semaglutide
6.4. Liraglutide
7. GLP-1RAs in the Pipeline
7.1. Mazdutide
7.2. Tirzepatide
8. Discussion
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yokose, C.; McCormick, N.; Lu, N.; Tanikella, S.; Lin, K.; Joshi, A.D.; Raffield, L.M.; Warner, E.; Merriman, T.; Hsu, J.; et al. Trends in Prevalence of Gout Among US Asian Adults, 2011–2018. JAMA Netw. Open 2023, 6, e239501. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, E. Reduced glomerular function and prevalence of gout: NHANES 2009–10. PLoS ONE 2012, 7, e50046. [Google Scholar] [CrossRef]
- McAdams-DeMarco, M.A.; Maynard, J.W.; Baer, A.N.; Coresh, J. Hypertension and the risk of incident gout in a population-based study: The atherosclerosis risk in communities cohort. J. Clin. Hypertens. 2012, 14, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Shi, W.; Zhang, N.; Tang, H.; Feng, X. Newer Glucose-Lowering Drugs and Risk of Gout: A Network Meta-Analysis of Randomized Outcomes Trials. Clin. Ther. 2024, 46, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wang, Y.; Fu, T.; Zhou, W.; Ge, X.; Sha, X.; Guo, J.; Dong, C.; Guo, G. Gout and risk of diabetes mellitus: Meta-analysis of observational studies. Psychol. Health Med. 2020, 25, 917–930. [Google Scholar] [CrossRef]
- Manoj, A.; Das, S.; Kunnath Ramachandran, A.; Alex, A.T.; Joseph, A. SGLT2 inhibitors, an accomplished development in field of medicinal chemistry: An extensive review. Future Med. Chem. 2020, 12, 1961–1990. [Google Scholar] [CrossRef]
- Hediger, M.A.; Rhoads, D.B. Molecular physiology of sodium-glucose cotransporters. Physiol. Rev. 1994, 74, 993–1026. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Norton, L.; Defronzo, R.A. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr. Rev. 2011, 32, 515–531. [Google Scholar] [CrossRef]
- Ferrannini, E.; Solini, A. SGLT2 inhibition in diabetes mellitus: Rationale and clinical prospects. Nat. Rev. Endocrinol. 2012, 8, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Ehrenkranz, J.R.; Lewis, N.G.; Kahn, C.R.; Roth, J. Phlorizin: A review. Diabetes Metab. Res. Rev. 2005, 21, 31–38. [Google Scholar] [CrossRef]
- Bebernitz, G. 7.13—Sodium–Glucose Cotransporters. In Comprehensive Medicinal Chemistry III; Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 491–511. [Google Scholar] [CrossRef]
- Muller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J. Family-B G-protein-coupled receptors. Genome Biol. 2001, 2, reviews3013.1. [Google Scholar] [CrossRef]
- Amori, R.E.; Lau, J.; Pittas, A.G. Efficacy and safety of incretin therapy in type 2 diabetes: Systematic review and meta-analysis. JAMA 2007, 298, 194–206. [Google Scholar] [CrossRef]
- Tanday, N.; Flatt, P.R.; Irwin, N. Metabolic responses and benefits of glucagon-like peptide-1 (GLP-1) receptor ligands. Br. J. Pharmacol. 2022, 179, 526–541. [Google Scholar] [CrossRef] [PubMed]
- Korner, M.; Stockli, M.; Waser, B.; Reubi, J.C. GLP-1 receptor expression in human tumors and human normal tissues: Potential for in vivo targeting. J. Nucl. Med. 2007, 48, 736–743. [Google Scholar] [CrossRef]
- Baggio, L.L.; Yusta, B.; Mulvihill, E.E.; Cao, X.; Streutker, C.J.; Butany, J.; Cappola, T.P.; Margulies, K.B.; Drucker, D.J. GLP-1 Receptor Expression Within the Human Heart. Endocrinology 2018, 159, 1570–1584. [Google Scholar] [CrossRef]
- Asmar, A.; Asmar, M.; Simonsen, L.; Madsbad, S.; Holst, J.J.; Hartmann, B.; Sorensen, C.M.; Bulow, J. Glucagon-like peptide-1 elicits vasodilation in adipose tissue and skeletal muscle in healthy men. Physiol. Rep. 2017, 5, e13073. [Google Scholar] [CrossRef] [PubMed]
- Asmar, A.; Cramon, P.K.; Simonsen, L.; Asmar, M.; Sorensen, C.M.; Madsbad, S.; Moro, C.; Hartmann, B.; Jensen, B.L.; Holst, J.J.; et al. Extracellular Fluid Volume Expansion Uncovers a Natriuretic Action of GLP-1: A Functional GLP-1-Renal Axis in Man. J. Clin. Endocrinol. Metab. 2019, 104, 2509–2519. [Google Scholar] [CrossRef]
- Skov, J.; Dejgaard, A.; Frokiaer, J.; Holst, J.J.; Jonassen, T.; Rittig, S.; Christiansen, J.S. Glucagon-like peptide-1 (GLP-1): Effect on kidney hemodynamics and renin-angiotensin-aldosterone system in healthy men. J. Clin. Endocrinol. Metab. 2013, 98, E664–E671. [Google Scholar] [CrossRef]
- Madaan, T.; Akhtar, M.; Najmi, A.K. Sodium glucose CoTransporter 2 (SGLT2) inhibitors: Current status and future perspective. Eur. J. Pharm. Sci. 2016, 93, 244–252. [Google Scholar] [CrossRef]
- Sarnoski-Brocavich, S.; Hilas, O. Canagliflozin (invokana), a novel oral agent for type-2 diabetes. Pharm. Ther. 2013, 38, 656–666. [Google Scholar]
- Chen, X.; Hu, P.; Vaccaro, N.; Polidori, D.; Curtin, C.R.; Stieltjes, H.; Sha, S.; Weiner, S.; Devineni, D. Pharmacokinetics, Pharmacodynamics, and Safety of Single-Dose Canagliflozin in Healthy Chinese Subjects. Clin. Ther. 2015, 37, 1483–1492. [Google Scholar] [CrossRef]
- Usiskin, K.; Kline, I.; Fung, A.; Mayer, C.; Meininger, G. Safety and tolerability of canagliflozin in patients with type 2 diabetes mellitus: Pooled analysis of phase 3 study results. Postgrad. Med. 2014, 126, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Trujillo, A.; Vijapurkar, U.; Damaraju, C.V.; Meininger, G. Effect of canagliflozin on serum uric acid in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2015, 17, 426–429. [Google Scholar] [CrossRef]
- Doi, Y.; Hamano, T.; Yamaguchi, S.; Sakaguchi, Y.; Kaimori, J.Y.; Isaka, Y. Mediators between canagliflozin and renoprotection vary depending on patient characteristics: Insights from the CREDENCE trial. Diabetes Obes. Metab. 2023, 25, 2944–2953. [Google Scholar] [CrossRef]
- Li, J.; Badve, S.V.; Zhou, Z.; Rodgers, A.; Day, R.; Oh, R.; Lee, M.; Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; et al. The effects of canagliflozin on gout in type 2 diabetes: A post-hoc analysis of the CANVAS Program. Lancet Rheumatol. 2019, 1, e220–e228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef]
- Sridharan, K.; Alkhidir, M.M.O.H. Hypouricemic effect of sodium glucose transporter-2 inhibitors: A network meta-analysis and meta-regression of randomized clinical trials. Expert Rev. Endocrinol. Metab. 2025, 20, 139–146. [Google Scholar] [CrossRef]
- Banerjee, M.; Pal, R.; Maisnam, I.; Chowdhury, S.; Mukhopadhyay, S. Serum uric acid lowering and effects of sodium-glucose cotransporter-2 inhibitors on gout: A meta-analysis and meta-regression of randomized controlled trials. Diabetes Obes. Metab. 2023, 25, 2697–2703. [Google Scholar] [CrossRef]
- Scheen, A.J. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin. Pharmacokinet. 2014, 53, 213–225. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Inzucchi, S.E.; Mattheus, M.; Meinicke, T.; Steubl, D.; Wanner, C.; Zinman, B. Empagliflozin and uric acid metabolism in diabetes: A post hoc analysis of the EMPA-REG OUTCOME trial. Diabetes Obes. Metab. 2022, 24, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Ji, Q.; Bhatt, D.L.; Mazer, C.D.; Al-Omran, M.; Inzucchi, S.E.; Wanner, C.; Ofstad, A.P.; Zwiener, I.; George, J.T.; et al. Association between uric acid levels and cardio-renal outcomes and death in patients with type 2 diabetes: A subanalysis of EMPA-REG OUTCOME. Diabetes Obes. Metab. 2020, 22, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Doehner, W.; Anker, S.D.; Butler, J.; Zannad, F.; Filippatos, G.; Ferreira, J.P.; Salsali, A.; Kaempfer, C.; Brueckmann, M.; Pocock, S.J.; et al. Uric acid and sodium-glucose cotransporter-2 inhibition with empagliflozin in heart failure with reduced ejection fraction: The EMPEROR-reduced trial. Eur. Heart J. 2022, 43, 3435–3446. [Google Scholar] [CrossRef]
- Bechlioulis, A.; Markozannes, G.; Chionidi, I.; Liberopoulos, E.; Naka, K.K.; Ntzani, E.E.; Liatis, S.; Rizzo, M.; Rizos, E.C. The effect of SGLT2 inhibitors, GLP1 agonists, and their sequential combination on cardiometabolic parameters: A randomized, prospective, intervention study. J. Diabetes Complications. 2023, 37, 108436. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Bohm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Doehner, W.; Anker, S.D.; Butler, J.; Zannad, F.; Filippatos, G.; Coats, A.J.S.; Ferreira, J.P.; Henrichmoeller, I.; Brueckmann, M.; Schueler, E.; et al. Uric Acid and SGLT2 Inhibition with Empagliflozin in Heart Failure with Preserved Ejection Fraction: The EMPEROR-Preserved Trial. JACC Heart Fail 2024, 12, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Mayne, K.J.; Sardell, R.J.; Staplin, N.; Judge, P.K.; Zhu, D.; Sammons, E.; Cherney, D.Z.I.; Green, J.B.; Levin, A.; Pontremoli, R.; et al. Empagliflozin lowers serum uric acid in chronic kidney disease: Exploratory analyses from the EMPA-KIDNEY trial. Nephrol. Dial Transpl. 2024, 40, 720–730. [Google Scholar] [CrossRef]
- Akbari, A.; Rafiee, M.; Sathyapalan, T.; Sahebkar, A. Impacts of Sodium/Glucose Cotransporter-2 Inhibitors on Circulating Uric Acid Concentrations: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2022, 2022, 7520632. [Google Scholar] [CrossRef]
- Hu, X.; Yang, Y.; Hu, X.; Jia, X.; Liu, H.; Wei, M.; Lyu, Z. Effects of sodium-glucose cotransporter 2 inhibitors on serum uric acid in patients with type 2 diabetes mellitus: A systematic review and network meta-analysis. Diabetes Obes. Metab. 2022, 24, 228–238. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Liu, X.; Lacreta, F.; Griffen, S.C.; Boulton, D.W. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin. Pharmacokinet. 2014, 53, 17–27. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- McDowell, K.; Welsh, P.; Docherty, K.F.; Morrow, D.A.; Jhund, P.S.; de Boer, R.A.; O’Meara, E.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; et al. Dapagliflozin reduces uric acid concentration, an independent predictor of adverse outcomes in DAPA-HF. Eur. J. Heart Fail. 2022, 24, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Butt, J.H.; Docherty, K.F.; Claggett, B.L.; Desai, A.S.; Petersson, M.; Langkilde, A.M.; de Boer, R.A.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Association of Dapagliflozin Use with Clinical Outcomes and the Introduction of Uric Acid-Lowering Therapy and Colchicine in Patients with Heart Failure with and Without Gout: A Patient-Level Pooled Meta-analysis of DAPA-HF and DELIVER. JAMA Cardiol. 2023, 8, 386–393. [Google Scholar] [CrossRef]
- Stack, A.G.; Han, D.; Goldwater, R.; Johansson, S.; Dronamraju, N.; Oscarsson, J.; Johnsson, E.; Parkinson, J.; Erlandsson, F. Dapagliflozin Added to Verinurad Plus Febuxostat Further Reduces Serum Uric Acid in Hyperuricemia: The QUARTZ Study. J. Clin. Endocrinol. Metab. 2021, 106, e2347–e2356. [Google Scholar] [CrossRef]
- Mulder, S.; Hammarstedt, A.; Nagaraj, S.B.; Nair, V.; Ju, W.; Hedberg, J.; Greasley, P.J.; Eriksson, J.W.; Oscarsson, J.; Heerspink, H.J.L. A metabolomics-based molecular pathway analysis of how the sodium-glucose co-transporter-2 inhibitor dapagliflozin may slow kidney function decline in patients with diabetes. Diabetes Obes. Metab. 2020, 22, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Behera, B.S.; Singh, S.; Munshi, A. The pharmacological profile of SGLT2 inhibitors: Focus on mechanistic aspects and pharmacogenomics. Eur. J. Pharmacol. 2021, 904, 174169. [Google Scholar] [CrossRef]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef]
- Seino, Y.; Sasaki, T.; Fukatsu, A.; Sakai, S.; Samukawa, Y. Efficacy and safety of luseogliflozin monotherapy in Japanese patients with type 2 diabetes mellitus: A 12-week, randomized, placebo-controlled, phase II study. Curr. Med. Res. Opin. 2014, 30, 1219–1230. [Google Scholar] [CrossRef]
- Seino, Y.; Sasaki, T.; Fukatsu, A.; Ubukata, M.; Sakai, S.; Samukawa, Y. Efficacy and safety of luseogliflozin as monotherapy in Japanese patients with type 2 diabetes mellitus: A randomized, double-blind, placebo-controlled, phase 3 study. Curr. Med. Res. Opin. 2014, 30, 1245–1255. [Google Scholar] [CrossRef]
- Haneda, M.; Seino, Y.; Inagaki, N.; Kaku, K.; Sasaki, T.; Fukatsu, A.; Kakiuchi, H.; Sato, Y.; Sakai, S.; Samukawa, Y. Influence of Renal Function on the 52-Week Efficacy and Safety of the Sodium Glucose Cotransporter 2 Inhibitor Luseogliflozin in Japanese Patients with Type 2 Diabetes Mellitus. Clin. Ther. 2016, 38, 66–88. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Kaku, K.; Seino, Y.; Inagaki, N.; Haneda, M.; Sasaki, T.; Fukatsu, A.; Kakiuchi, H.; Samukawa, Y. Efficacy and Safety of the SGLT2 Inhibitor Luseogliflozin in Japanese Patients with Type 2 Diabetes Mellitus Stratified According to Baseline Body Mass Index: Pooled Analysis of Data from 52-Week Phase III Trials. Clin. Ther. 2016, 38, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, A.; Kazuta, K.; Yoshida, S.; Nagase, I. Randomized, placebo-controlled, double-blind glycemic control trial of novel sodium-dependent glucose cotransporter 2 inhibitor ipragliflozin in Japanese patients with type 2 diabetes mellitus. J. Diabetes Investig. 2014, 5, 382–391. [Google Scholar] [CrossRef]
- Kawata, T.; Iizuka, T.; Iemitsu, K.; Takihata, M.; Takai, M.; Nakajima, S.; Minami, N.; Umezawa, S.; Kanamori, A.; Takeda, H.; et al. Ipragliflozin Improves Glycemic Control and Decreases Body Fat in Patients with Type 2 Diabetes Mellitus. J. Clin. Med. Res. 2017, 9, 586–595. [Google Scholar] [CrossRef]
- Tanaka, M.; Yamakage, H.; Inoue, T.; Odori, S.; Kusakabe, T.; Shimatsu, A.; Satoh-Asahara, N. Beneficial Effects of Ipragliflozin on the Renal Function and Serum Uric Acid Levels in Japanese Patients with Type 2 Diabetes: A Randomized, 12-week, Open-label, Active-controlled Trial. Intern. Med. 2020, 59, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Terauchi, Y.; Tamura, M.; Senda, M.; Gunji, R.; Kaku, K. Efficacy and safety of tofogliflozin in Japanese patients with type 2 diabetes mellitus with inadequate glycaemic control on insulin therapy (J-STEP/INS): Results of a 16-week randomized, double-blind, placebo-controlled multicentre trial. Diabetes Obes. Metab. 2017, 19, 1397–1407. [Google Scholar] [CrossRef]
- Kaku, K.; Watada, H.; Iwamoto, Y.; Utsunomiya, K.; Terauchi, Y.; Tobe, K.; Tanizawa, Y.; Araki, E.; Ueda, M.; Suganami, H.; et al. Efficacy and safety of monotherapy with the novel sodium/glucose cotransporter-2 inhibitor tofogliflozin in Japanese patients with type 2 diabetes mellitus: A combined Phase 2 and 3 randomized, placebo-controlled, double-blind, parallel-group comparative study. Cardiovasc. Diabetol. 2014, 13, 65. [Google Scholar] [CrossRef]
- Nguyen, V.K.; White, J.R., Jr. Overview of Ertugliflozin. Clin. Diabetes 2019, 37, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, V.S.; Cosentino, F.; Dagogo-Jack, S.; McGuire, D.K.; Pratley, R.E.; Cater, N.B.; Noyes Essex, M.; Mancuso, J.P.; Zhao, Y.; Cherney, D.Z.I. Effects of ertugliflozin on uric acid and gout-related outcomes in persons with type 2 diabetes and cardiovascular disease: Post hoc analyses from VERTIS CV. Diabetes Obes. Metab. 2024, 26, 5336–5346. [Google Scholar] [CrossRef]
- Najafi, S.; Bahrami, M.; Butler, A.E.; Sahebkar, A. The effect of glucagon-like peptide-1 receptor agonists on serum uric acid concentration: A systematic review and meta-analysis. Br. J. Clin. Pharmacol. 2022, 88, 3627–3637. [Google Scholar] [CrossRef]
- Nielsen, L.L.; Young, A.A.; Parkes, D.G. Pharmacology of exenatide (synthetic exendin-4): A potential therapeutic for improved glycemic control of type 2 diabetes. Regul. Pept. 2004, 117, 77–88. [Google Scholar] [CrossRef]
- Deacon, C.F.; Knudsen, L.B.; Madsen, K.; Wiberg, F.C.; Jacobsen, O.; Holst, J.J. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia 1998, 41, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Muskiet, M.H.; Tonneijck, L.; Smits, M.M.; Kramer, M.H.; Diamant, M.; Joles, J.A.; van Raalte, D.H. Acute renal haemodynamic effects of glucagon-like peptide-1 receptor agonist exenatide in healthy overweight men. Diabetes Obes. Metab. 2016, 18, 178–185. [Google Scholar] [CrossRef]
- Tonneijck, L.; Muskiet, M.H.A.; Smits, M.M.; Bjornstad, P.; Kramer, M.H.H.; Diamant, M.; Hoorn, E.J.; Joles, J.A.; van Raalte, D.H. Effect of immediate and prolonged GLP-1 receptor agonist administration on uric acid and kidney clearance: Post-hoc analyses of four clinical trials. Diabetes Obes. Metab. 2018, 20, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Tonneijck, L.; Smits, M.M.; Muskiet, M.H.A.; Hoekstra, T.; Kramer, M.H.H.; Danser, A.H.J.; Diamant, M.; Joles, J.A.; van Raalte, D.H. Acute renal effects of the GLP-1 receptor agonist exenatide in overweight type 2 diabetes patients: A randomised, double-blind, placebo-controlled trial. Diabetologia 2016, 59, 1412–1421. [Google Scholar] [CrossRef]
- Edwards, A.; Auberson, M.; Ramakrishnan, S.K.; Bonny, O. A model of uric acid transport in the rat proximal tubule. Am. J. Physiol.-Renal Physiol. 2019, 316, F934–F947. [Google Scholar] [CrossRef] [PubMed]
- Dutour, A.; Abdesselam, I.; Ancel, P.; Kober, F.; Mrad, G.; Darmon, P.; Ronsin, O.; Pradel, V.; Lesavre, N.; Martin, J.C.; et al. Exenatide decreases liver fat content and epicardial adipose tissue in patients with obesity and type 2 diabetes: A prospective randomized clinical trial using magnetic resonance imaging and spectroscopy. Diabetes Obes. Metab. 2016, 18, 882–891. [Google Scholar] [CrossRef]
- Sanford, M. Dulaglutide: First global approval. Drugs 2014, 74, 2097–2103. [Google Scholar] [CrossRef]
- Geiser, J.S.; Heathman, M.A.; Cui, X.; Martin, J.; Loghin, C.; Chien, J.Y.; de la Pena, A. Clinical Pharmacokinetics of Dulaglutide in Patients with Type 2 Diabetes: Analyses of Data from Clinical Trials. Clin. Pharmacokinet. 2016, 55, 625–634. [Google Scholar] [CrossRef]
- Acosta-Calero, C.; Arnas-Leon, C.; Santana-Suarez, A.D.; Nivelo-Rivadeneira, M.; Kuzior, A.; Quintana-Arroyo, S.; Sablon-Gonzalez, N.; Martinez-Martin, F.J. Dulaglutide added on Empagliflozin improves blood pressure, body weight, glycemic control and albuminuria in obese diabetic patients. Endocr. Abstr. 2017, 49, EP621. [Google Scholar] [CrossRef]
- Iwasaki, T.; Kessoku, T.; Higurashi, T.; Taguri, M.; Yoneda, M. Low body mass index and old age are useful in predicting the hemoglobin A1c-lowering effect of switching from sitagliptin to dulaglutide in Japanese patients with type 2 diabetes mellitus: A single-center, open-label, single-arm, pilot study. Diabetol. Int. 2018, 9, 189–195. [Google Scholar] [CrossRef]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Choudhary, N.S.; Singh, M.K.; Wasir, J.S.; Kaur, P.; Gill, H.K.; Bano, T.; Farooqui, K.J.; et al. Effect of dulaglutide on liver fat in patients with type 2 diabetes and NAFLD: Randomised controlled trial (D-LIFT trial). Diabetologia 2020, 63, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.T.; Baekdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Ronne, J.; Madsen, K.G.; Scheele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018, 10, eaar7047. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schaffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Tschop, M.H.; Friedman, J.M. Seeking satiety: From signals to solutions. Sci. Transl. Med. 2023, 15, eadh4453. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Yokokawa, H.; Nagamine, T.; Fukuda, H.; Hisaoka, T.; Naito, T. Efficacy and safety of semaglutide in glycemic control, body weight management, lipid profiles and other biomarkers among obese type 2 diabetes patients initiated or switched to semaglutide from other GLP-1 receptor agonists. J. Diabetes Metab. Disord. 2021, 20, 2121–2128. [Google Scholar] [CrossRef]
- Xiang, J.; Ding, X.Y.; Zhang, W.; Zhang, J.; Zhang, Y.S.; Li, Z.M.; Xia, N.; Liang, Y.Z. Clinical effectiveness of semaglutide on weight loss, body composition, and muscle strength in Chinese adults. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 9908–9915. [Google Scholar] [CrossRef]
- Reppo, I.; Jakobson, M.; Volke, V. Effects of Semaglutide and Empagliflozin on Inflammatory Markers in Patients with Type 2 Diabetes. Int. J. Mol. Sci. 2023, 24, 5714. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. 2019, 10, 155. [Google Scholar] [CrossRef]
- Friedman, J.M. The discovery and development of GLP-1 based drugs that have revolutionized the treatment of obesity. Proc. Natl. Acad. Sci. USA 2024, 121, e2415550121. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.A.; Chuang, S.F. Evaluation of the efficacy of low-dose liraglutide in weight control among Taiwanese non-diabetes patients. J. Diabetes Investig. 2020, 11, 1524–1531. [Google Scholar] [CrossRef]
- Molero, I.G.; Arnés, J.G.; Lopez, M.D.; Vallejo, R.; Gonzalo, M. Efficacy and Safety Evaluation of Liraglutide Treatment in Morbid Obese and Diabetic Patients in First Year of Commercialization in Spain. Diabetes Technol. 2013, 15, A125. [Google Scholar]
- Hiramatsu, T.; Asano, Y.; Mabuchi, M.; Imai, K.; Iguchi, D.; Furuta, S. Liraglutide relieves cardiac dilated function than DPP-4 inhibitors. Eur. J. Clin. Investig. 2018, 48, e13007. [Google Scholar] [CrossRef]
- Hiramatsu, T.; Ito, H.; Okumura, S.; Asano, Y.; Iguchi, D.; Furuta, S. Impact of glucagon like peptide-1 receptor agonist and sodium glucose cotransporter 2 inhibitors on type 2 diabetes patients with renal impairment. Diabetes Vasc. Dis. Res. 2020, 17, 1479164120971220. [Google Scholar] [CrossRef]
- Nakaguchi, H.; Kondo, Y.; Kyohara, M.; Konishi, H.; Oiwa, K.; Terauchi, Y. Effects of liraglutide and empagliflozin added to insulin therapy in patients with type 2 diabetes: A randomized controlled study. J. Diabetes Investig. 2020, 11, 1542–1550. [Google Scholar] [CrossRef]
- Ishii, H.; Niiya, T.; Ono, Y.; Inaba, N.; Jinnouchi, H.; Watada, H. Improvement of quality of life through glycemic control by liraglutide, a GLP-1 analog, in insulin-naive patients with type 2 diabetes mellitus: The PAGE1 study. Diabetol. Metab. Syndr. 2017, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Ticinovic Kurir, T.; Milicevic, T.; Novak, A.; Vilovic, M.; Bozic, J. Adropin-Potential Link in Cardiovascular Protection for Obese Male Type 2 Diabetes Mellitus Patients Treated with Liraglutide. Acta Clin. Croat. 2020, 59, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Liakos, A.; Lambadiari, V.; Bargiota, A.; Kitsios, K.; Avramidis, I.; Kotsa, K.; Gerou, S.; Boura, P.; Tentolouris, N.; Dimitriadis, G.; et al. Effect of liraglutide on ambulatory blood pressure in patients with hypertension and type 2 diabetes: A randomized, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 2019, 21, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Zobbe, K.; Nielsen, S.M.; Christensen, R.; Overgaard, A.; Gudbergsen, H.; Henriksen, M.; Bliddal, H.; Dreyer, L.; Stamp, L.; Knop, F.K.; et al. OP0172 Effect of Weight Loss and Liraglutide on Serum Urate Levels among Obese Knee Osteoarthritis Patients: Secondary Analysis of a Randomised Controlled Trial. Ann. Rheum. Dis. 2020, 79, 107–108. [Google Scholar] [CrossRef]
- Ji, L.; Jiang, H.; Cheng, Z.; Qiu, W.; Liao, L.; Zhang, Y.; Li, X.; Pang, S.; Zhang, L.; Chen, L.; et al. A phase 2 randomised controlled trial of mazdutide in Chinese overweight adults or adults with obesity. Nat. Commun. 2023, 14, 8289. [Google Scholar] [CrossRef]
- Ji, L.; Gao, L.; Jiang, H.; Yang, J.; Yu, L.; Wen, J.; Cai, C.; Deng, H.; Feng, L.; Song, B.; et al. Safety and efficacy of a GLP-1 and glucagon receptor dual agonist mazdutide (IBI362) 9 mg and 10 mg in Chinese adults with overweight or obesity: A randomised, placebo-controlled, multiple-ascending-dose phase 1b trial. EClinicalMedicine 2022, 54, 101691. [Google Scholar] [CrossRef]
- JI, L.; Jiang, H.; Li, H.; Tian, J.; Liu, D.; Zhao, Y.; Gu, J.; Liu, Z.; Deng, H.; Wang, Y.; et al. 1856-LB: Efficacy and Safety of Mazdutide in Chinese Participants with Overweight or Obesity (GLORY-1). Diabetes 2024, 73, 1856-LB. [Google Scholar] [CrossRef]
- Ji, L.; Jiang, H.; An, P.; Deng, H.; Liu, M.; Li, L.; Feng, L.; Song, B.; Han-Zhang, H.; Ma, Q.; et al. IBI362 (LY3305677), a weekly-dose GLP-1 and glucagon receptor dual agonist, in Chinese adults with overweight or obesity: A randomised, placebo-controlled, multiple ascending dose phase 1b study. EClinicalMedicine 2021, 39, 101088. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.C.; Hojlund, M.; Henriksen, D.P.; Hallas, J.; Kristensen, K.B. Sodium-glucose cotransporter-2 inhibitors and the risk of gout: A Danish population based cohort study and symmetry analysis. Pharmacoepidemiol. Drug Saf. 2021, 30, 1391–1395. [Google Scholar] [CrossRef]
- Fralick, M.; Chen, S.K.; Patorno, E.; Kim, S.C. Assessing the Risk for Gout with Sodium-Glucose Cotransporter-2 Inhibitors in Patients with Type 2 Diabetes: A Population-Based Cohort Study. Ann. Intern. Med. 2020, 172, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Alsalem, A.; Alsultan, M.M.; Alqarni, F.; Almangour, A.; Alsharekh, L.; Alenazi, S.; Alzahrani, S.; Almanqour, R.A.; Alazmi, A.; Alzahrani, A. Real-world evidence of the effects of sodium-glucose co-transporter 2 inhibitors on the dosing of diuretics in patients with heart failure: A retrospective cohort study. Front. Pharmacol. 2024, 15, 1366439. [Google Scholar] [CrossRef]
- Sridharan, K. Possible attenuation of gout risk by SGLT-2 inhibitors in patients using thiazide and loop diuretics. Expert Rev. Endocrinol. Metab. 2025, 20, 249–250. [Google Scholar] [CrossRef]
- Sun, H.L.; Wu, Y.W.; Bian, H.G.; Yang, H.; Wang, H.; Meng, X.M.; Jin, J. Function of Uric Acid Transporters and Their Inhibitors in Hyperuricaemia. Front. Pharmacol. 2021, 12, 667753. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takahashi, M.; Yan, K.; Sakurai, H. Expression of SLC2A9 isoforms in the kidney and their localization in polarized epithelial cells. PLoS ONE 2014, 9, e84996. [Google Scholar] [CrossRef]
- Novikov, A.; Fu, Y.; Huang, W.; Freeman, B.; Patel, R.; van Ginkel, C.; Koepsell, H.; Busslinger, M.; Onishi, A.; Nespoux, J.; et al. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol.-Renal Physiol. 2019, 316, F173–F185. [Google Scholar] [CrossRef]
- Chakrabarti, K.; McCune, W.J. SGLT-2 inhibitors: New horizons for rheumatologists. Curr. Opin. Rheumatol. 2024, 36, 351–359. [Google Scholar] [CrossRef]
- Komiya, C.; Tsuchiya, K.; Shiba, K.; Miyachi, Y.; Furuke, S.; Shimazu, N.; Yamaguchi, S.; Kanno, K.; Ogawa, Y. Ipragliflozin Improves Hepatic Steatosis in Obese Mice and Liver Dysfunction in Type 2 Diabetic Patients Irrespective of Body Weight Reduction. PLoS ONE 2016, 11, e0151511. [Google Scholar] [CrossRef] [PubMed]
- Basalay, M.V.; Davidson, S.M.; Yellon, D.M. Neuroprotection in Rats Following Ischaemia-Reperfusion Injury by GLP-1 Analogues-Liraglutide and Semaglutide. Cardiovasc. Drugs Ther. 2019, 33, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, L.A.; Mankad, S.; Sokos, G.G.; Miske, G.; Shah, A.; Elahi, D.; Shannon, R.P. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004, 109, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Yusta, B.; Baggio, L.L.; Koehler, J.; Holland, D.; Cao, X.; Pinnell, L.J.; Johnson-Henry, K.C.; Yeung, W.; Surette, M.G.; Bang, K.A.; et al. GLP-1R Agonists Modulate Enteric Immune Responses Through the Intestinal Intraepithelial Lymphocyte GLP-1R. Diabetes 2015, 64, 2537–2549. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Name of Drug | Gout Incidence | SUA | Study Design | Participants | Study Duration | References |
|---|---|---|---|---|---|---|
| Canagliflozin | Reduced | Post hoc analysis of four phase III studies | T2DM patients (n = 115) | 100 or 300 mg/day administered for 26 weeks | [25] | |
| Reduced | Post hoc analysis of the CREDENCE study | T2DM patients with kidney disease (n = 153, 224 in the canagliflozin and placebo respectively) | 100 mg administered for 52 weeks | [26] | ||
| Reduced | Post hoc analysis of CANVAS AND CANVAS-Renal studies | T2DM patients elevated CVD risk (n = 10,142) | 100 or 300 mg/day up to 7 years (CANVAS) and 3.6 years (CANVAS-R) | [27] | ||
| Empagliflozin | Reduced gout flares | Reduced | A randomized, double-blind, placebo-controlled study. EMPA-REG OUTCOME trial. | T2DM patients with established atherosclerotic CVD (n = 7020) | 10 and 25 mg/day up to 3 years. | [32] |
| Reduced gout flares, gouty arthritis, and initiation of gout therapy | Reduced | A phase III randomized, double-blind parallel-group, placebo-controlled trial (EMPEROR-Reduced trial) | T2DM patients with heart failure (n = 3676) | 10 mg/day. 4–100 weeks | [34] | |
| Reduced | A randomized prospective intervention study comparing liraglutide with empagliflozin | T2DM patients (25% with established CVD disease) (n = 62) | 25 mg/day for 3 months | [35] | ||
| Reduced acute gout, gouty arthritis, and initiation of gout therapy | Reduced | Post hoc analysis of EMPEROR-Preserved (a phase III double-blind, parallel-group, placebo-controlled, event-driven trial) | chronic heart failure patients with preserved ejection fraction (n = 5924) | 10 mg/day up to 172 weeks | [36,37] | |
| Did not reduce the risk of gout | Reduced | Post hoc analysis of EMPA-KIDNEY (a phase III double-blind, parallel-group, placebo-controlled trial) | Patients with chronic kidney disease (n = 6609) | 10 mg/day up to 18 months | [38] | |
| Dapagliflozin | Reduced initiation of gout therapy. No information about gout flares | Reduced | Follow-up on the DAPA-HF study (phase III, placebo-controlled, randomized) | Patients with heart failure and reduced ejection fraction with and without T2DM (n = 3119) | 10 mg/day for 52 weeks | [43] |
| Reduced initiation of gout therapy and colchicine therapy | Post hoc analysis of DAPA-HF and DELIVER trials | [44] | ||||
| Reduced | Quartz study (randomized, placebo-controlled crossover study) | Adults with asymptomatic hyperuricemia (n = 36) | combination of verinurad (9 mg/day) and febuxostat (80 mg/day) with and without Dapagliflozin (10 mg/day) for 7 days | [45] | ||
| Luseogliflozin | Reduced | Single- and multiple-dose trial | Healthy subjects (n = 57, 24 for single and multiple doses respectively) | single dose (1–25 mg/day) and 7-day multiple dose | [48] | |
| Reduced | Phase II randomized, placebo-controlled, double-blind study | T2DM patients (n = 232) | 0.5, 2.5 and 5 mg/day for 12 weeks | [49] | ||
| Reduced | Phase III randomized, double-blind, placebo-controlled, parallel-group comparative study | T2DM patients (n = 148) | 2.5 mg/day for 24 weeks | [50] | ||
| Reduced | Pooled analysis of four phase III studies | T2DM with different renal impairment or BMI | 2.5 mg/day (or up to 5 mg/day) for 52 weeks | [51] | ||
| Ipragliflozin | No change from baseline | Phase II double-blind, multicenter, placebo-controlled dose-response study | T2DM Japanese patients (n = 360) | 12.5, 25, 50 or 100 mg/day) for 12 weeks | [53] | |
| Reduced | ASSIGN-K | T2DM patients (n = 367) | 50 mg/day for 24 weeks | [54] | ||
| Reduced | Randomized, open-label, active-controlled small trial | Inadequately controlled T2DM patients (n = 30) | 50 mg/day for 12 weeks | [55] | ||
| Tofogliflozin | Reduced | Phase IV multicenter double-blind, placebo-controlled trial | Inadequately controlled T2DM patients (n = 211) | 20 mg/day for 16 weeks (and 36 weeks open label extension) | [56] | |
| Reduced | Combined phase II and III randomized, placebo-controlled, double-blind, multicenter, parallel-group study | T2DM patients (n = 229) | 10, 20 and 40 mg/day for 24 weeks | [57] | ||
| Ertugliflozin | Reduced incidence of gout events | Reduced | VERTIS CV—phase III, multicenter, double-blind, placebo-controlled | T2DM patients with atherosclerotic and cardiovascular disease (n = 8246) | 5 and 15 mg/day up to 260 weeks | [59] |
| SUA | Clinical Data | Participants | Design | References | |
|---|---|---|---|---|---|
| Exenatide | Increased but also increased absolute UA excretion | Post hoc analysis | Healthy overweight subjects (n = 9) | Acute infusion of 10 µg exenatide for 150 min following 90-min placebo infusion | [63,64] |
| No change | Acute randomized, double-blind, placebo-controlled, parallel-group study | T2DM patients (n = 52) | 10 µg infusion | [64,65] | |
| No change | Prospective randomized clinical study | Obese individuals with T2DM (n = 44) | Obese individuals with T2DM receive 5 µg exenatide twice daily for 4 weeks followed by 10 μg twice daily until week 26 | [67] | |
| Dulaglutide | Reduced | Open observational study | Patients with T2DM (n = 20) previously treated with empagliflozin (10 mg/day) | Dulaglutide administration at 1.5 mg/week for 3–6 months | [70] |
| No change | Open-label, parallel-group, randomized, controlled study | Patients with T2DM and non-alcoholic fatty liver disease (n = 64) | 24-week administration of dulaglutide (0.75 mg/week for 4 weeks, 1.5 mg/week for 20 weeks) | [71] | |
| No change | Single-center, open-label, pilot study | T2DM patients previously treated with 50 mg/day sitagliptin (n = 40) | 24-week dulaglutide administration (0.75 mg/week) | [72] | |
| Semaglutide | Reduced | Retrospective cohort study | T2DM patients (n = 50) | Semaglutide was administered 0.25 mg/week and increased to 0.5 or 1 mg/week after 4 weeks for 3–6 months | [76] |
| Reduced | Retrospective study | Chinese participants with obesity (n = 43) | Semaglutide administered at 0.25 mg/week increased every two weeks up to 1.0 mg/week for 24 weeks | [77] | |
| No change | Prospective clinical trial comparing semaglutide and empagliflozide | T2DM patients (n = 20) | Three-month administration of semaglutide (0.25 mg/week s.c. increase to 0.5 mg/week on week 5 and to 1 mg/week on week 9) | [78] | |
| Liraglutide | Reduced | Retrospective study | Non-diabetic obese patients with metabolic syndrome (n = 46) | 12-week administration of liraglutode 0.6 or 1.2 mg/day | [81] |
| Reduced | Retrospective study | T2DM obese patients (n = 54) | Not mentioned | [82] | |
| No change | Longitudinal study comparing liraglutide vs. sitagliptin and linagliptin | T2DM patients with renal impairment (n = 139) | Liraglutide (0.9 mg/day) vs. sitagliptin (50 mg/day) and linagliptin (5 mg/day) administered for 48 months | [83] | |
| Reduced less than dapagliflozin and empagliflozin | Comparison study between liraglutide (0.9 mg/day) and dapagliflozin (5 mg/day) or empagliflozin (10 mg/day) | T2DM patients with renal impairment (n = 188) | Liraglutide administered 0.9 mg/day for 36 months | [84] | |
| No change | Open-label, parallel-group, randomized, controlled trial comparing liraglutide to empagliflozin combined with insulin therapy | T2DM patients naive to GLP-1RA or DPP4i treatment | 24-week liraglutide (0.9 mg/day) vs. empagliflozin (10 mg/day) administration | [85] | |
| Reduced | Prospective, multicenter, observational study | T2DM patients (n = 151) | Liraglutide administration starting at 0.3 mg/day and increased to 0.9 mg/day for 12 weeks | [86] | |
| No change | Non-randomized, controlled, interventional study | T2DM obese patients (n = 15) | Liraglutide (0.6 mg/day titrated to 1.2 mg/day after 2 weeks) administered for 3 months | [87] | |
| No change | Parallel-group, randomized, double-blind, placebo-controlled study | T2DM patients (n = 62) | Liraglutide (0.6 mg/day titrated to 1.2 mg/day after 1 weeks) administered for 5 weeks | [88] | |
| Reduced | Randomized, double-blind, placebo-controlled, parallel-group study (LOSE-IT) | Obese individuals with knee osteoarthritis | 8-week intensive Cambridge weight management plan + 52 weeks liraglutide (3 mg/day) | [89] | |
| Mazdutide | Reduced | Randomized, double-blind placebo-controlled trial (GLORY-1) | Overweight or obese Chinese adults (n = 610) | Mazdutide administered at 4 mg/week and 6 mg/week vs. placebo for 48 weeks | [92] |
| Reduced | Randomized, double-blind, placebo-controlled phase 2 study | Overweight or obese Chinese adults (n = 248) | Mazdutide administered at 3, 4.5, and 6 mg/week for 24 weeks | [90] | |
| Reduced | Randomized, placebo-controlled, dose-escalation, multiple ascending dose phase 1b study | Overweight or obese Chinese adults (n = 12) | Mazdutide administered at 3, 4.5 and 6 mg/week for 12 weeks | [93] | |
| Reduced | Randomized, placebo-controlled, multiple ascending dose, phase 1b study | Overweight or obese Chinese adults (n = 24) | Mazdutide administered for 12 weeks at 9 mg/week target dose and 16 weeks at 10 mg/week target dose | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaufmann, D.; Schlesinger, N. Could Sodium-Glucose Co-Transporter-2 Inhibitors and Glucagon-like Peptide-1 Receptor Agonists Play a Role in Gout Treatment? Pharmaceutics 2025, 17, 865. https://doi.org/10.3390/pharmaceutics17070865
Kaufmann D, Schlesinger N. Could Sodium-Glucose Co-Transporter-2 Inhibitors and Glucagon-like Peptide-1 Receptor Agonists Play a Role in Gout Treatment? Pharmaceutics. 2025; 17(7):865. https://doi.org/10.3390/pharmaceutics17070865
Chicago/Turabian StyleKaufmann, Dan, and Naomi Schlesinger. 2025. "Could Sodium-Glucose Co-Transporter-2 Inhibitors and Glucagon-like Peptide-1 Receptor Agonists Play a Role in Gout Treatment?" Pharmaceutics 17, no. 7: 865. https://doi.org/10.3390/pharmaceutics17070865
APA StyleKaufmann, D., & Schlesinger, N. (2025). Could Sodium-Glucose Co-Transporter-2 Inhibitors and Glucagon-like Peptide-1 Receptor Agonists Play a Role in Gout Treatment? Pharmaceutics, 17(7), 865. https://doi.org/10.3390/pharmaceutics17070865