
Development and Characterization of a New Oral Antileishmanial Bis(pyridine-2-Carboxamidine) Drug Through Innovative Dissolution Testing in Biorelevant Media Combined with Pharmacokinetic Studies

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
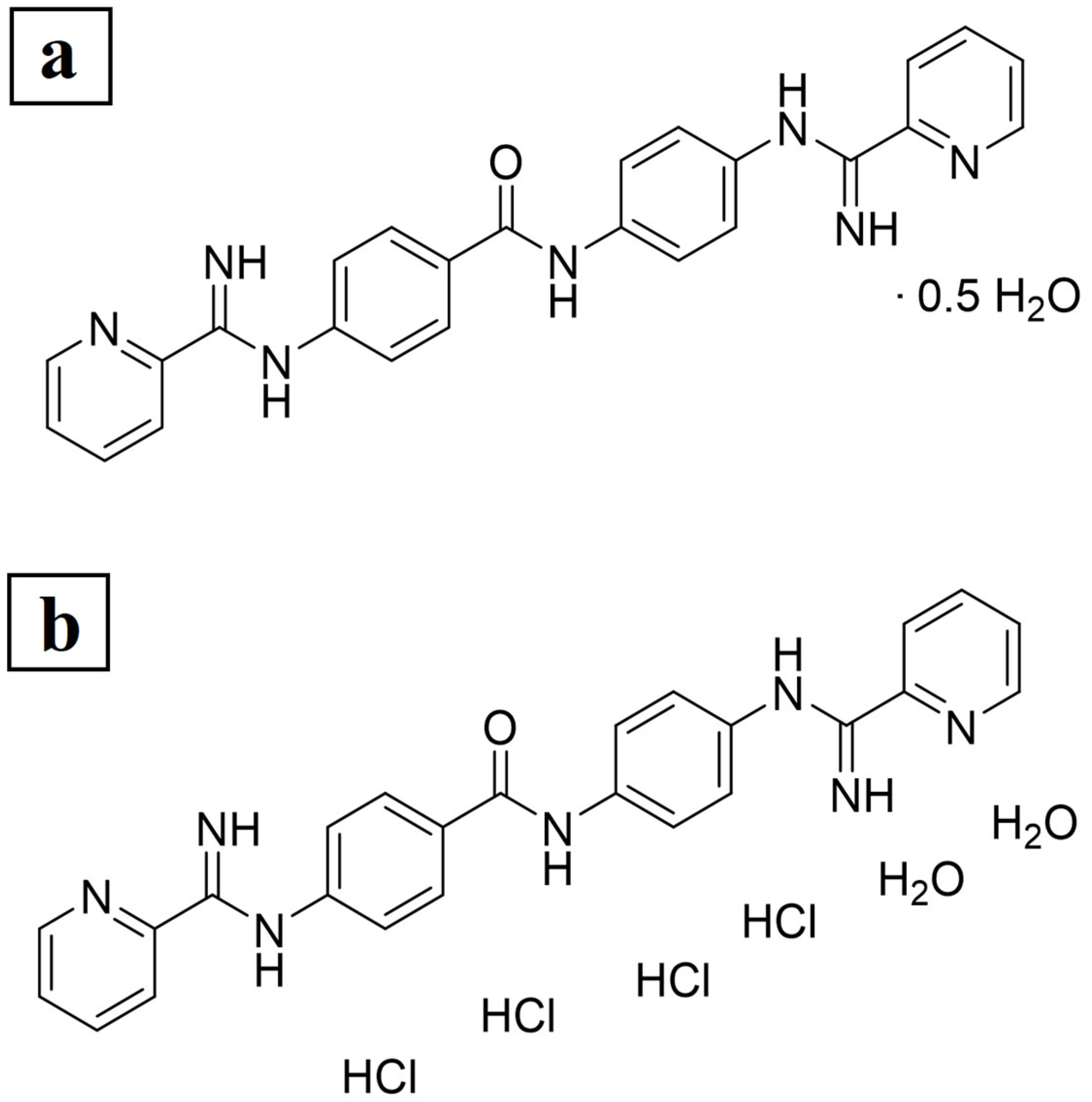
2.2.1. Preparation of JNII40_base and JNII40_HCl Formulations
2.2.2. Solid State Characterization
Elemental Analysis (EA)
Differential Scanning Calorimetry (DSC)
Fourier Transform Infrared Study (FTIR)
Scanning Electron Microscopy (SEM)
2.2.3. Dissolution Studies
2.2.4. Solubility Studies
2.2.5. Animal Study
2.2.6. Plasma Concentration-Time Profile
2.2.7. HPLC-MS/MS Analytical Method for Pharmacokinetics
2.2.8. Bioavailability Parameters
2.2.9. Statistics Analysis
3. Results and Discussion
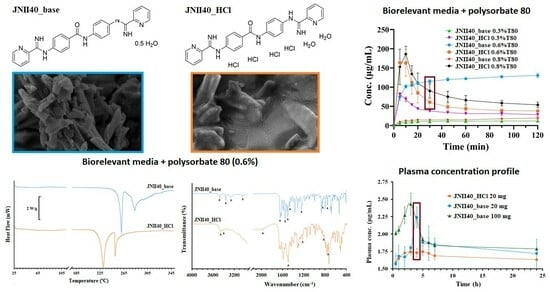
3.1. Characterization at the Solid State
3.1.1. Elemental Analysis
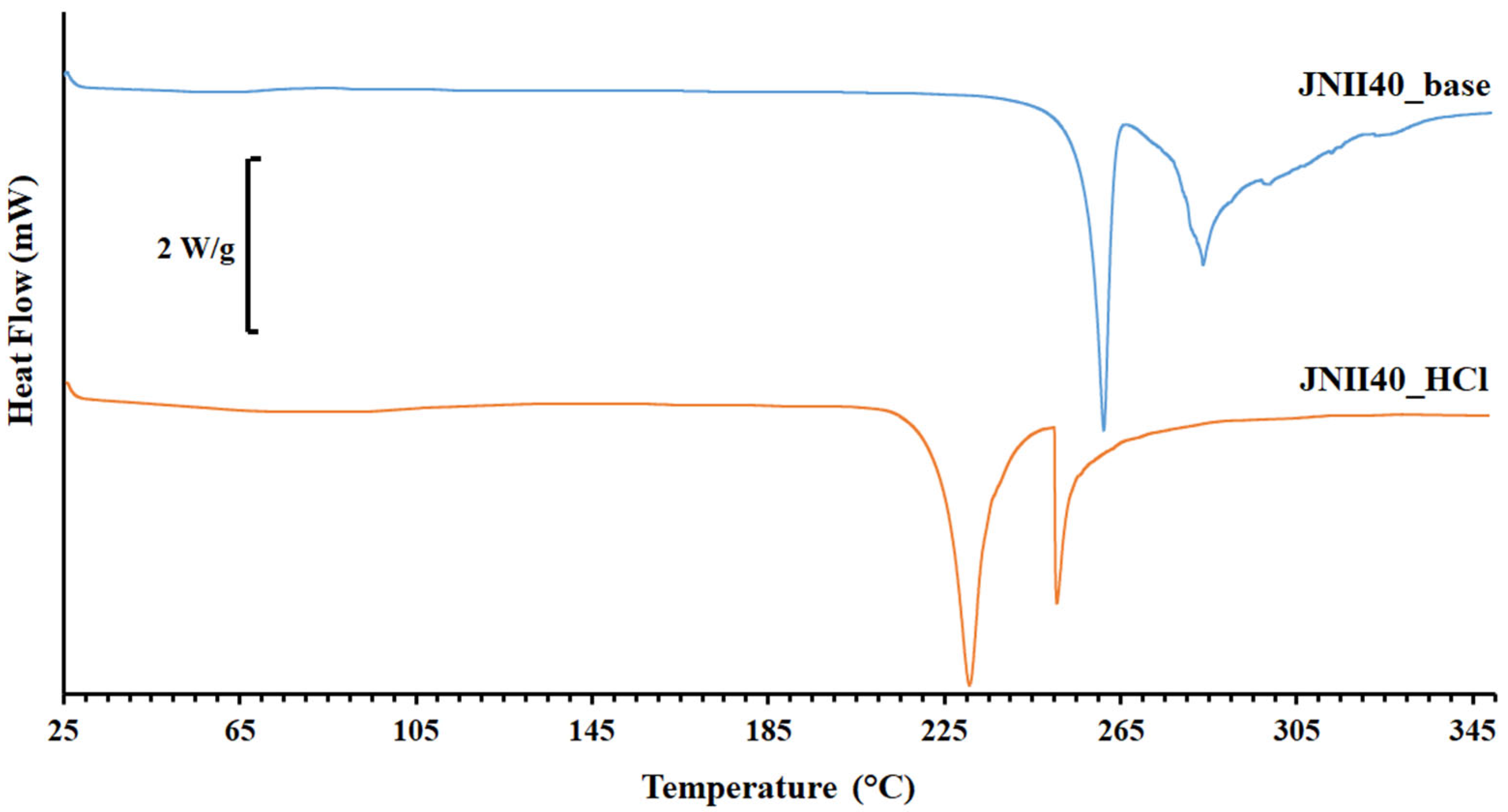
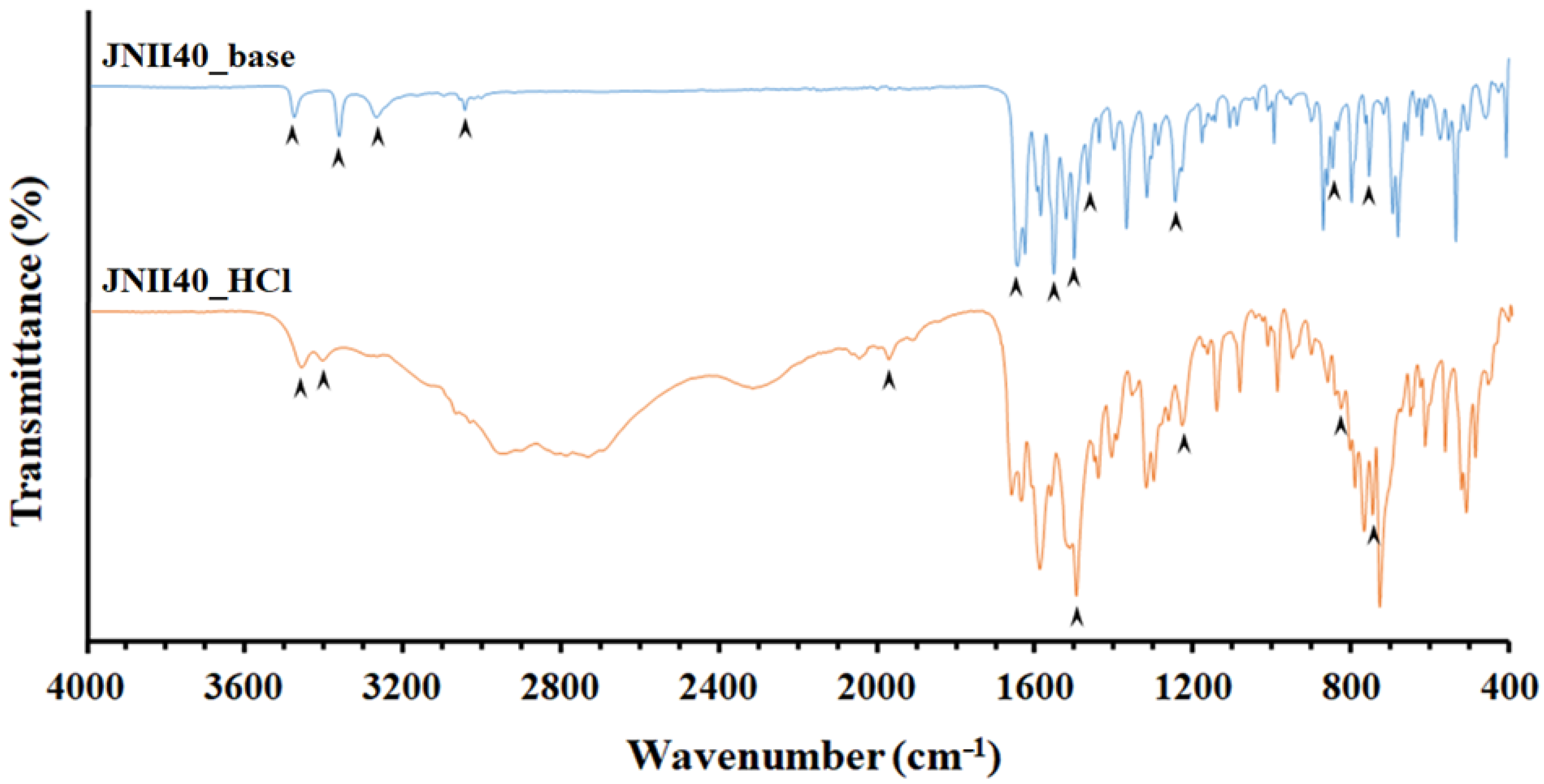
3.1.2. Differential Scanning Calorimetry (DSC) and Fourier Transform Infrared Study (FTIR)
3.1.3. Scanning Electron Microscopy (SEM) Characterization
3.2. Dissolution Studies
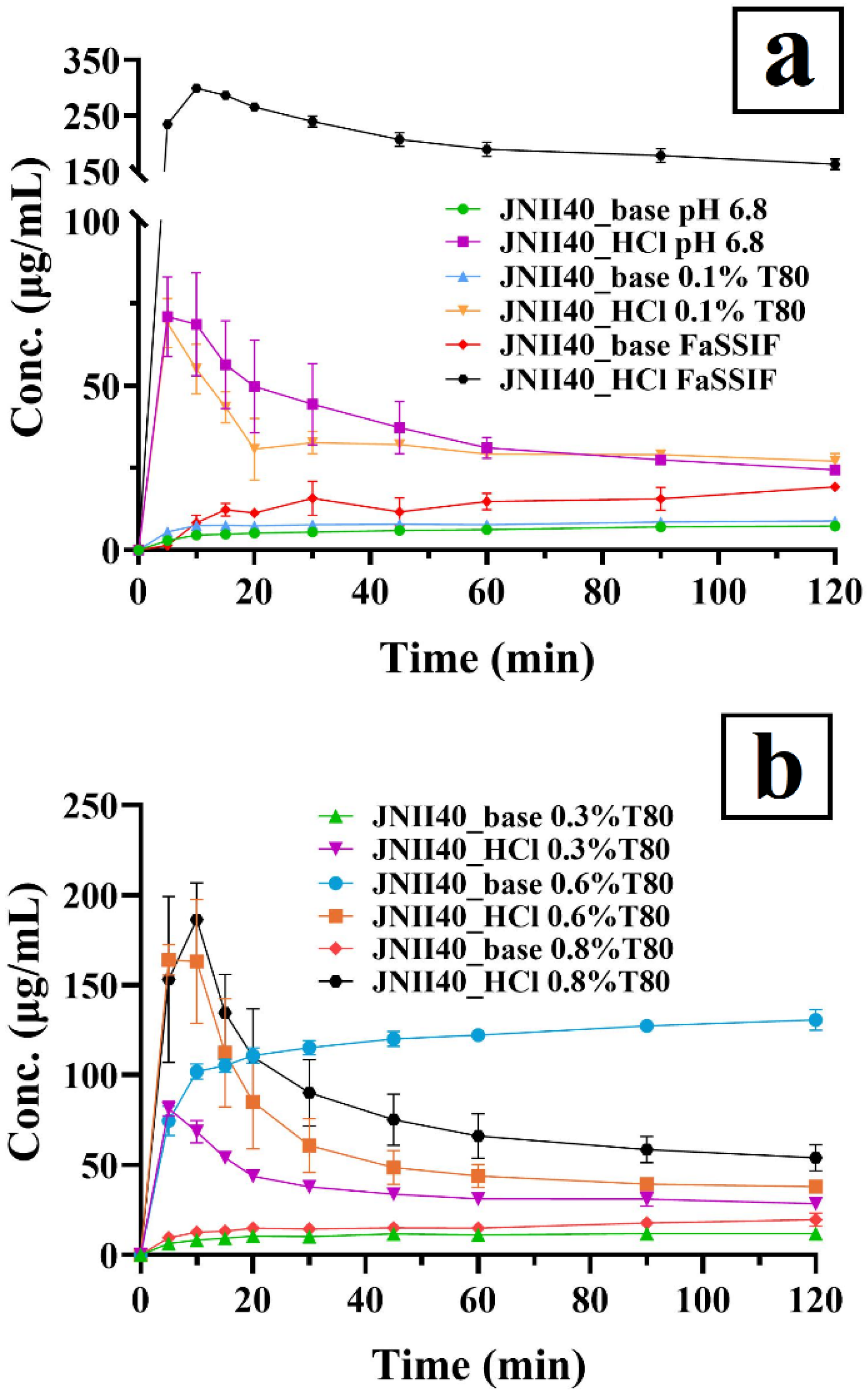
3.2.1. Dissolution Studies in Different Intestinal Simulated Media
3.2.2. Dissolution Studies in Biorelevant Media at Different Concentrations of Polysorbate 80
3.3. Solubility Studies in Biorelevant Media: Influence of Ionic Strength and Polysorbate 80 Proportions
3.4. Pharmacokinetic Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NTD | Neglected tropical disease |
| CL | Cutaneous leishmaniasis |
| VL | Visceral leishmaniasis |
| IC50 | Inhibitory concentration 50 |
| IC90 | Inhibitory concentration 90 |
| JNII40_base | Name of the base compound: 4-picolinimidamido-N-[4-(picolinimidamido)phenyl]benzamide |
| JNII40_HCl | Hydrochloride salt form of the JNII40 compound |
| FaSSGF | Fasted State Simulated Gastric Fluid |
| FaSSIF | Fasted State Simulated Intestinal Fluid |
| SDS | Sodium lauryl sulfate |
| SNEDDS | Self-Nanoemulsifying Drug Delivery Systems |
| PBS | Phosphate buffered saline |
| SLN | Solid lipid nanoparticle |
| DSPS | Dissolution-perfusion system |
| P407 | Poloxamer 407 |
| TPGS | Tocopherol Polyethylene Glycol Succinate |
| EA | Elemental analysis |
| DSC | Differential scanning calorimetry |
| FTIR | Fourier transform infrared spectroscopy |
| SEM | Scanning electron microscopy |
| HPLC-MS/MS | High-performance liquid chromatography–mass spectrometry |
| UV-VIS | Ultraviolet–visible spectrophotometry |
| 1H NMR | Proton nuclear magnetic resonance |
| LC-MS | Liquid chromatography–mass spectrometry |
| MRM | Multiple reaction monitoring |
| ESI | Electrospray ionization |
| SPE | Solid-phase extraction |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| Cmax | Maximum plasma concentration |
| Tmax | Time to maximum plasma concentration |
| AUC0–24h | Area under the curve from 0 to 24 h |
| ANOVA | Analysis of variance |
| SE | Standard error |
| Tg | Glass transition |
| SD | Standard deviation |
| C24h | Plasma concentration at 24 h |
| Log p | Partition coefficient |
| t1/2 | Half-life |
References
- Singh, V.K.; Tiwari, R.; Rajneesh, N.; Kumar, A.; Chauhan, S.B.; Sudarshan, M.; Mehrotra, S.; Gautam, V.; Sundar, S.; Kumar, R. Advancing Treatment for Leishmaniasis: From Overcoming Challenges to Embracing Therapeutic Innovations. ACS Infect. Dis. 2025, 11, 47–68. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Leishmaniasis. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 1 April 2024).
- Sindermann, H.; Engel, J. Development of miltefosine as an oral treatment for leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, S17–S20. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Souza, F.; da Silva, V.D.; Silva, G.X.; Taniwaki, N.N.; Hardoim, D.d.J.; Buarque, C.D.; Abreu-Silva, A.L.; Calabrese, K.d.S. 1,4-disubstituted-1,2,3-triazole compounds induce ultrastructural alterations in leishmania amazonensis promastigote: An in vitro antileishmanial and in silico pharmacokinetic study. Int. J. Mol. Sci. 2020, 21, 6839. [Google Scholar] [CrossRef]
- Jiménez-Antón, M.D.; García-Calvo, E.; Gutiérrez, C.; Escribano, M.D.; Kayali, N.; Luque-García, J.L.; Olías-Molero, A.I.; Corral, M.J.; Costi, M.P.; Torrado, J.J.; et al. Pharmacokinetics and disposition of miltefosine in healthy mice and hamsters experimentally infected with Leishmania infantum. Eur. J. Pharm. Sci. 2018, 121, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Verrest, L.; Roseboom, I.C.; Wasunna, M.; Mbui, J.; Njenga, S.; Musa, A.M.; Olobo, J.; Mohammed, R.; Ritmeijer, K.; Chu, W.Y.; et al. Population pharmacokinetics of a combination of miltefosine and paromomycin in Eastern African children and adults with visceral leishmaniasis. J. Antimicrob. Chemother. 2023, 78, 2702–2714. [Google Scholar] [CrossRef]
- Palić, S.; Beijnen, J.H.; Dorlo, T.P.C. An update on the clinical pharmacology of miltefosine in the treatment of leishmaniasis. Int. J. Antimicrob. Agents 2022, 59, 106459. [Google Scholar] [CrossRef]
- Gogou, G.; Koutsoni, O.S.; Stathopoulos, P.; Skaltsounis, L.A.; Halabalaki, M.; Dotsika, E. Direct In Vitro Comparison of the Anti-Leishmanial Activity of Different Olive Oil Total Polyphenolic Fractions and Assessment of Their Combined Effects with Miltefosine. Molecules 2022, 27, 6176. [Google Scholar] [CrossRef]
- Gedda, M.R.; Madhukar, P.; Vishwakarma, A.K.; Verma, V.; Kushwaha, A.K.; Yadagiri, G.; Mudavath, S.L.; Singh, O.P.; Srivastava, O.N.; Sundar, S. Evaluation of Safety and Antileishmanial Efficacy of Amine Functionalized Carbon-Based Composite Nanoparticle Appended with Amphotericin B: An in vitro and Preclinical Study. Front. Chem. 2020, 8, 510. [Google Scholar] [CrossRef]
- Vitorino, L.S.; dos Santos, T.C.; Bessa, I.A.A.; Santos, E.C.S.; Verçoza, B.R.F.; de Oliveira, L.A.S.; Rodrigues, J.C.F.; Ronconi, C.M. Amphotericin-B-loaded polymer-functionalized reduced graphene oxides for Leishmania amazonensis chemo-photothermal therapy. Colloids Surf. B Biointerfaces 2022, 209 Pt 1, 112169. [Google Scholar] [CrossRef]
- Smith, L.; Serrano, D.R.; Mauger, M.; Bolás-Fernández, F.; Dea-Ayuela, M.A.; Lalatsa, A. Orally Bioavailable and Effective Bu-parvaquone Lipid-Based Nanomedicines for Visceral Leishmaniasis. Mol. Pharm. 2018, 15, 2570–2583. [Google Scholar] [CrossRef]
- Machín, L.; Nápoles, R.; Gille, L.; Monzote, L. Leishmania amazonensis response to artemisinin and derivatives. Parasitol. Int. 2021, 80, 102218. [Google Scholar] [CrossRef]
- Kim, H.S.; Ortiz, D.; Kadayat, T.M.; Fargo, C.M.; Hammill, J.T.; Chen, Y.; Rice, A.L.; Begley, K.L.; Shoeran, G.; Pistel, W.; et al. Optimization of Orally Bioavailable Antileishmanial 2,4,5-Trisubstituted Benzamides. J. Med. Chem. 2023, 66, 7374–7386. [Google Scholar] [CrossRef]
- Etxebeste-Mitxeltorena, M.; Moreno, E.; Carvalheiro, M.; Calvo, A.; Navarro-Blasco, I.; González-Peñas, E.; Álvarez-Galindo, J.I.; Plano, D.; Irache, J.M.; Almeida, A.J.; et al. Oral Efficacy of a Diselenide Compound Loaded in Nanostructured Lipid Carriers in a Murine Model of Visceral Leishmaniasis. ACS Infect. Dis. 2021, 7, 3197–3209. [Google Scholar] [CrossRef] [PubMed]
- Nué-Martinez, J.J.; Cisneros, D.; Moreno-Blázquez, M.d.V.; Fonseca-Berzal, C.; Manzano, J.I.; Kraeutler, D.; Ungogo, M.A.; Aloraini, M.A.; Elati, H.A.A.; Ibáñez-Escribano, A.; et al. Synthesis and Biophysical and Biological Studies of N-Phenylbenzamide Derivatives Targeting Kinetoplastid Parasites. J. Med. Chem. 2023, 66, 13452–13480. [Google Scholar] [CrossRef] [PubMed]
- Devarapalli, R.; Indukuri, A.; Bollineni, M.; Mondal, A.; Reddy, C.M.; Chennuru, R. Investigation of Poor Solubility of a Salt-Cocrystal Hydrate: A Case Study of the Common-Ion Effect in Betrixaban, an Anticoagulant Drug. Mol. Pharm. 2021, 18, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, I.I.; Uçar, O. The Hirshfeld Surface Analysis of Antimony(III) and Bismuth(III) Complexes with Dithiocarbamate Ligands. J. Balk. Sci. Technol. 2022, 1, 87–95. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Tang, M.J.; Ho, N.J.; Shek, T.L.; Yang, Z.; Zuo, Z. Screening of Bioequivalent Extended-Release Formulations for Metformin by Principal Component Analysis and Convolution-Based IVIVC Approach. AAPS J. 2021, 23, 38. [Google Scholar] [CrossRef]
- Öztürk, K.; Arslan, F.B.; Öztürk, S.C.; Çalış, S. Mixed micelles formulation for carvedilol delivery: In-vitro characterization and in-vivo evaluation. Int. J. Pharm. 2022, 611, 121294. [Google Scholar] [CrossRef]
- Saha, S.K.; Joshi, A.; Singh, R.; Jana, S.; Dubey, K. An investigation into solubility and dissolution improvement of alectinib hydrochloride as a third-generation amorphous solid dispersion. J. Drug Deliv. Sci. Technol. 2023, 81, 104259. [Google Scholar] [CrossRef]
- Thakore, S.D.; Sirvi, A.; Joshi, V.C.; Panigrahi, S.S.; Manna, A.; Singh, R.; Sangamwar, A.T.; Bansal, A.K. Biorelevant dissolution testing and physiologically based absorption modeling to predict in vivo performance of supersaturating drug delivery systems. Int. J. Pharm. 2021, 607, 120958. [Google Scholar] [CrossRef]
- Arnold, Y.E.; Imanidis, G.; Kuentz, M.T. Advancing in-vitro drug precipitation testing: New process monitoring tools and a kinetic nucleation and growth model. J. Pharm. Pharmacol. 2011, 63, 333–341. [Google Scholar] [CrossRef]
- Higashino, H.; Minami, K.; Takagi, T.; Kataoka, M.; Yamashita, S. The effects of degree and duration of supersaturation on in vivo absorption profiles for highly permeable drugs, dipyridamole and ketoconazole. Eur. J. Pharm. Biopharm. 2023, 189, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Naing, M.D.; Tsume, Y. Dissolution profiles of BCS class II drugs generated by the gastrointestinal simulator alpha has an edge over the compendial USP II method. Eur. J. Pharm. Biopharm. 2024, 203, 114436. [Google Scholar] [CrossRef]
- García-Herrero, V.; Torrado-Salmerón, C.; García-Rodríguez, J.J.; Torrado, G.; Torrado-Santiago, S. Submicellar liquid chromatography with fluorescence detection improves the analysis of naproxen in plasma and brain tissue. J. Sep. Sci. 2019, 42, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Dong, X. Design and Evaluation of Two-Step Biorelevant Dissolution Methods for Docetaxel Oral Formulations. AAPS PharmSciTech 2022, 23, 113. [Google Scholar] [CrossRef] [PubMed]
- Singhal, M.; Turunen, E.; Ahtola-Sätilä, T.; Aspegren, J.; Bratty, J.R.; Fuhr, R.; Ojala, K.; van Veen, B.; Peltonen, L. Nanoparticle-based oral formulation can surprise you with inferior in vivo absorption in humans. Eur. J. Pharm. Biopharm. 2022, 177, 91–99. [Google Scholar] [CrossRef]
- Tak, J.W.; Kwon, T.K.; Kim, Y.I.; Cho, J.H.; Kim, J.; Kim, J.O. Development of abiraterone acetate tablets with enhanced oral bioavailability. J. Pharm. Investig. 2024, 54, 345–356. [Google Scholar] [CrossRef]
- Chen, G.; Zhu, Y.; Wang, Q.; Bai, Y.; Ma, S.; Wang, J.; Zhao, M.; Zou, M.; Cheng, G. The development of a novel simultaneous in vitro dissolution—In situ perfusion system as a potential tool for studying the absorption of solid oral formulation in rat. Eur. J. Pharm. Sci. 2023, 191, 106601. [Google Scholar] [CrossRef]
- Dwivedi, P.; Khatik, R.; Khandelwal, K.; Taneja, I.; Raju, K.S.R.; Wahajuddin; Paliwal, S.K.; Dwivedi, A.K.; Mishra, P.R. Pharmacokinetics study of arteether loaded solid lipid nanoparticles: An improved oral bioavailability in rats. Int. J. Pharm. 2014, 466, 321–327. [Google Scholar] [CrossRef]
- Yukuyama, M.N.; Zuo, J.; Park, C.; Yousef, M.; Henostroza, M.A.B.; de Araujo, G.L.B.; Bou-Chacra, N.A.; Löbenberg, R. Biphasic dissolution combined with modified cylinder method—A new promising method for dissolution test in drug-loaded nanoemulsions. Int. J. Pharm. 2023, 632, 122554. [Google Scholar] [CrossRef]
- Huang, Z.; Parikh, S.; Fish, W.P. Interactions between a poorly soluble cationic drug and sodium dodecyl sulfate in dissolution medium and their impact on in vitro dissolution behavior. Int. J. Pharm. 2018, 535, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Yang, S.G. Enhanced oral bioavailability of megestrol acetate via polymer-hybridized solid lipid nanoparticles using ultrasonic nebulization method. J. Pharm. Investig. 2024, 55, 105–112. [Google Scholar] [CrossRef]
- Aldeeb, R.A.; El-Miligi, M.F.; El-Nabarawi, M.; Tag, R.; Amin, H.M.S.; Taha, A.A. Enhancement of the solubility and dissolution rate of telmisartan by surface solid dispersions employing superdisintegrants, hydrophilic polymers and combined carriers. Sci. Pharm. 2022, 90, 71. [Google Scholar] [CrossRef]
- Li, Y.W.; Zhang, H.M.; Cui, B.J.; Hao, C.Y.; Zhu, H.Y.; Guan, J.; Wang, D.; Jin, Y.; Feng, B.; Cai, J.; et al. “Felodipine-indomethacin” co-amorphous supersaturating drug delivery systems: “Spring-parachute” process, stability, in vivo bioavailability, and underlying molecular mechanisms. Eur. J. Pharm. Biopharm. 2021, 166, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Torrado-Salmerón, C.; Guarnizo-Herrero, V.; Torrado, G.; Peña, M.Á.; Torrado-Santiago, S.; de la Torre-Iglesias, P.M. Solid dispersions of atorvastatin with Kolliphor RH40: Enhanced supersaturation and improvement in a hyperlipidemic rat model. Int. J. Pharm. 2023, 631, 122520. [Google Scholar] [CrossRef]
- Xu, J.; Qiao, H.; Gan, L.; Wang, P.; Wang, J.; Cui, Y.; Zhou, J.; Liu, Q.; Jiang, Y.; Zhang, H.; et al. Zinc caproate: Ecofriendly synthesis, structural characterization, and antibacterial action. Int. J. Pharm. 2024, 655, 124030. [Google Scholar] [CrossRef]
- Pereira, J.A.; Diniz, L.F.; Lehmann, C.W.; Carvalho, P.S. Pharmaceutical salts of carvedilol with increased solubility as an approach for enhancing the drug action. Cryst. Growth Des. 2024, 24, 6658–6672. [Google Scholar] [CrossRef]
- Nugrahani, I.; Parwati, R.D. Challenges and progress in nonsteroidal anti-inflammatory drugs co-crystal development. Molecules 2021, 26, 4185. [Google Scholar] [CrossRef]
- Hall, A.V.; Gostick, I.E.F.; Yufit, D.S.; Marchant, G.Y.; Kirubakaran, P.; Madu, S.J.; Li, M.; Steel, P.G.; Steed, J.W. Integral role of water in the solid-state behavior of the antileishmanial drug miltefosine. Cryst. Growth Des. 2022, 22, 6262–6266. [Google Scholar] [CrossRef]
- Huang, Z.; Staufenbiel, S.; Bodmeier, R. Combination of co-crystal and nanocrystal techniques to improve the solubility and dissolution rate of poorly soluble drugs. Pharm. Res. 2022, 39, 949–961. [Google Scholar] [CrossRef]
- Jia, S.; Ning, S.; Leng, Y.; Jing, Q.; Xu, Z.; Ren, F. Stabilizing effect of Soluplus on erlotinib metastable crystal form in microparticles and amorphous solid dispersions. Polymers 2022, 14, 1241. [Google Scholar] [CrossRef] [PubMed]
- López-Manzanara Pérez, C.; Torres-Pabón, N.S.; Laguna, A.; Torrado, G.; de la Torre-Iglesias, P.M.; Torrado-Santiago, S.; Torrado-Salmerón, C. Development of chitosan/sodium carboxymethylcellulose complexes to improve the simvastatin release rate: Polymer/polymer and drug/polymer interactions’ effects on kinetic models. Polymers 2023, 15, 4184. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Yadagiri, G.; Negi, M.; Kushwaha, A.K.; Singh, O.P.; Sundar, S.; Mudavath, S.L. Carboxymethyl chitosan modified lipid nanoformulations as a highly efficacious and biocompatible oral anti-leishmanial drug carrier system. Int. J. Biol. Macromol. 2022, 204, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.S.; Rao, A.V.; Prasad, M.S.N.A.; Viswanath, I.V.K.; Laxminarayana, E. Some new 1,2,4-triazole derivatives bearing the pyrimidine moiety as potential antimycobacterial agents: Synthesis and docking analysis. Lett. Drug Des. Discov. 2022, 20, 1664–1674. [Google Scholar] [CrossRef]
- Liu, S.; Yue, K.; Qian, J.; Lu, D.; Wu, P.; Li, Q.; Zhang, Z. Integrated approach for improving mechanical and high-temperature properties of fast-growing poplar wood using lignin-controlled treatment combined with densification. Int. J. Biol. Macromol. 2024, 280 Pt 2, 135949. [Google Scholar] [CrossRef]
- Simin, A.; Ghaffarifar, F.; Delavari, H.; Dayer, M.S.; Hamidianfar, N.; Baghkhani, F. In vitro and in vivo effects of ethanolic extract of Fumaria parviflora Lam. embedded in chitosan nanoparticles against Leishmania major. Acta Parasitol. 2024, 69, 628–638. [Google Scholar] [CrossRef]
- Terashima, H.; Ozeki, T. The impact of sinkers on coning issues exhibited by tablets in USP2 dissolution apparatus. Int. J. Pharm. 2024, 659, 124236. [Google Scholar] [CrossRef]
- Kim, J.S.; Park, H.; Kang, K.T.; Ha, E.S.; Kim, M.S.; Hwang, S.J. Micronization of a poorly water-soluble drug, fenofibrate, via supercritical-fluid-assisted spray-drying. J. Pharm. Investig. 2022, 52, 353–366. [Google Scholar] [CrossRef]
- O’Dwyer, P.J.; Box, K.J.; Imanidis, G.; Vertzoni, M.; Reppas, C. On the usefulness of four in vitro methods in assessing the intraluminal performance of poorly soluble, ionisable compounds in the fasted state. Eur. J. Pharm. Sci. 2022, 168, 106034. [Google Scholar] [CrossRef]
- Navarro-Ruíz, E.; Álvarez-Álvarez, C.; Peña, M.Á.; Torrado-Salmerón, C.; Dahma, Z.; de la Torre-Iglesias, P.M. Multiparticulate Systems of Meloxicam for Colonic Administration in Cancer or Autoimmune Diseases. Pharmaceutics 2022, 14, 1504. [Google Scholar] [CrossRef]
- Di Gregorio, M.C.; Cautela, J.; Galantini, L. Physiology and Physical Chemistry of Bile Acids. Int. J. Mol. Sci. 2021, 22, 1780. [Google Scholar] [CrossRef] [PubMed]
- Dhaval, M.; Dudhat, K.; Gadoya, A.; Shah, S.; Pethani, T.; Jambukiya, N.; Patel, A.; Kalsariya, C.; Ansari, J.; Borkhataria, C. Pharmaceutical Salts: Comprehensive Insights From Fundamental Chemistry to FDA Approvals (2019–2023). AAPS PharmSciTech 2025, 26, 36. [Google Scholar] [CrossRef] [PubMed]
- Van Duong, T.; Ni, Z.; Taylor, L.S. Phase Behavior and Crystallization Kinetics of a Poorly Water-Soluble Weakly Basic Drug as a Function of Supersaturation and Media Composition. Mol. Pharm. 2022, 19, 1146–1159. [Google Scholar] [CrossRef]
- Nué-Martinez, J.J.; Leo-Barriga, M.; Herranz, F.; Koutsogiannis, Z.; Denny, P.W.; Ebiloma, G.U.; Dardonville, C.; González-Paredes, A. Nanostructured lipid carrier for intracellular delivery of a bis(pyridine-2-carboxamidine) DNA minor groove binder active against Leishmania. ACS Omega 2025, 10, 7795–7805. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.O.; Molnar, V.; Freijs, A.; Nygren, P.; Bergh, J.; Larsson, R. Pharmacokinetic models for the saturable distribution of paclitaxel. Drug Metab. Dispos. 1999, 27, 1220–1223. [Google Scholar] [CrossRef]
- Hakami, T.; Mahmoud, M.I.; de Juan, E.; Cooney, M. Pharmacokinetics of genistein distribution in blood and retinas of diabetic and non-diabetic rats. Drug Metab. Pharmacokinet. 2021, 39, 100404. [Google Scholar] [CrossRef]
- Anwer, M.K.; Iqbal, M.; Aldawsari, M.F.; Alalaiwe, A.; Ahmed, M.M.; Muharram, M.M.; Ezzeldin, E.; Mahmoud, M.A.; Imam, F.; Ali, R. Improved antimicrobial activity and oral bioavailability of delafloxacin by self-nanoemulsifying drug delivery system (SNEDDS). J. Drug Deliv. Sci. Technol. 2021, 64, 102572. [Google Scholar] [CrossRef]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Ferreira, R.A.A.; Junior, C.d.O.R.; Martinez, P.D.G.; Koovits, P.J.; Soares, B.M.; Ferreira, L.L.G.; Michelan-Duarte, S.; Chelucci, R.C.; Andricopulo, A.D.; Galuppo, M.K.; et al. 2-aminobenzimidazoles for leishmaniasis: From initial hit discovery to in vivo profiling. PLoS Negl. Trop. Dis. 2021, 15, e0009196. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Formulation | Media | Supersaturation Time (Increase-Fold) | Process | Ref. |
|---|---|---|---|---|
| Dipyridamole Ketoconazole | FaSSIF 1 pH 6.5 | Supersaturation 30 min; Precipitation dipyridamole 60–120 min and ketoconazole 30–60 min | Precipitation was related to a crystallization process | Higashino et al., 2023 [23] |
| Buparvaquone SNEDDS 3 | Lipolysis medium pH 6.5 bile salts and phosphatidylcholine | Supersaturation 60–80 min; No precipitation process | Bile salts and lipase suspension promote Lypolysis of SNEDDS | Smith et al., 2018 [11] |
| Dipyridamole | Two state FaSSGF 2 to FaSSIF 1 | Supersaturation 20 min; Precipitation 20–180 min | Precipitation was related to a crystallization process | Arnold et al., 2011 [22] |
| Telmisartan Solid dispersion | 1.5% SDS 6 in phosphate buffer pH 7.4 | Supersaturation 20–60 min ~3.5–4.0-fold; No precipitation process | Increase wettability and available surface area | Aldeeb et al., 2022 [34] |
| Arteether SLN 4 (2% polysorbate 80) | 0.1% polysorbate 80 in PBS pH 7.4 | Supersaturation at 50 min ~1.7-fold; No precipitation process | Increase wettability and available surface area | Dwivedi et al., 2014 [30] |
| Niterdipine micronized tablets (2.5% SDS 6) | DSPS 5 (Krebs-Ringer buffer pH 6.8) | Supersaturation at 30 min ~1.17-fold; Precipitation 30–240 min | Permeability dependent on pre-dissolved drug solution | Chen et al., 2023 [29] |
| Drug B tablets (poorly soluble cationic drug) | 0.5% SDS 6 in sodium acetate buffer pH 4.5 | Supersaturation at 60 min ~4.0-fold; No precipitation process | Micelle facilitated dissolution; Less soluble dodecyl sulfate salt of Drug B | Huang et al., 2018 [32] |
| Ritonavir | FaSSIF 1 pH 6.8 | Supersaturation at 30 min Precipitation 30–60 min | Bile acid increases solubilization | Naing et al., 2024 [24] |
| Albendazole:Soluplus 1:5 Solid dispersions (P407 5%) | FaSSIF 1 | Supersaturation 45–90 min ~2.0-fold; No precipitation process | Improve wettability and prevent drug recrystallization | Saha et al., 2023 [20] |
| Docetaxel granules (Mygliol 812: TPGS 1:1) | 0.5% polysorbate 80 PBS pH 7.4 | Supersaturation 10–240 min ~2.5-fold; No precipitation process | Micelle-facilitated dissolution | Shah et al., 2022 [26] |
| ODM-106 nanosuspension (0.1% SDS 6) | 0.5% polysorbate 80 phosphate buffer pH 6.8 non-sink condition | Supersaturation 0–30 min >2-fold; Precipitation process 60–60 min | Polysorbate 80 increase wettability | Singhal et al., 2022 [27] |
| Felodipine Co-amorphous systems | 0.25% SDS 6 in water solution | Supersaturation 30–60 min 1.53~1.87-fold; No precipitation process | Amorphization increased wettability and available surface area | Li et al., 2021 [35] |
| Plasma Parameters | JNII40_Base 100 mg/kg | JNII40_Base 20 mg/kg | JNII40_HCl 20 mg/kg |
|---|---|---|---|
| Cmax (µg/mL) | 2.42 ± 0.26 | 2.24 ± 0.22 | 1.75 ± 0.15 |
| Tmax (h) | 3.00 ± 0.00 | 4.00 ± 0.00 | 3.40 ± 0.89 |
| C24h (µg/mL) | 1.77 ± 0.23 | 1.68 ± 0.18 | 1.63 ± 0.22 |
| AUC0–24h (µg h/mL) | 44.68 ± 3.68 | 41.69 ± 4.29 | 39.32 ± 5.02 |
| Plasma Parameters | JNII40_Base 100 mg/kg | JNII40_Base 20 mg/kg |
|---|---|---|
| Promastigote | ||
| Cmax | 21.4 × IC50 | 19.8 × IC50 |
| C24h | 15.7 × IC50 | 14.9 × IC50 |
| t > 6 × IC50 | 24 h | 24 h |
| Amastigote | ||
| Cmax | 8.5 × IC50 | 7.9 × IC50 |
| C24h | 6.3 × IC50 | 6.0 × IC50 |
| t > 6 × IC50 | 23.5 h | 21 h |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laguna, A.; Martínez-Alonso, B.; Guarnizo-Herrero, V.; Nué-Martinez, J.J.; Dardonville, C.; Torrado-Santiago, S.; Torrado-Salmerón, C. Development and Characterization of a New Oral Antileishmanial Bis(pyridine-2-Carboxamidine) Drug Through Innovative Dissolution Testing in Biorelevant Media Combined with Pharmacokinetic Studies. Pharmaceutics 2025, 17, 838. https://doi.org/10.3390/pharmaceutics17070838
Laguna A, Martínez-Alonso B, Guarnizo-Herrero V, Nué-Martinez JJ, Dardonville C, Torrado-Santiago S, Torrado-Salmerón C. Development and Characterization of a New Oral Antileishmanial Bis(pyridine-2-Carboxamidine) Drug Through Innovative Dissolution Testing in Biorelevant Media Combined with Pharmacokinetic Studies. Pharmaceutics. 2025; 17(7):838. https://doi.org/10.3390/pharmaceutics17070838
Chicago/Turabian StyleLaguna, Almudena, Borja Martínez-Alonso, Víctor Guarnizo-Herrero, J. Jonathan Nué-Martinez, Christophe Dardonville, Santiago Torrado-Santiago, and Carlos Torrado-Salmerón. 2025. "Development and Characterization of a New Oral Antileishmanial Bis(pyridine-2-Carboxamidine) Drug Through Innovative Dissolution Testing in Biorelevant Media Combined with Pharmacokinetic Studies" Pharmaceutics 17, no. 7: 838. https://doi.org/10.3390/pharmaceutics17070838
APA StyleLaguna, A., Martínez-Alonso, B., Guarnizo-Herrero, V., Nué-Martinez, J. J., Dardonville, C., Torrado-Santiago, S., & Torrado-Salmerón, C. (2025). Development and Characterization of a New Oral Antileishmanial Bis(pyridine-2-Carboxamidine) Drug Through Innovative Dissolution Testing in Biorelevant Media Combined with Pharmacokinetic Studies. Pharmaceutics, 17(7), 838. https://doi.org/10.3390/pharmaceutics17070838