

Droplet Size Reduction of Self-Emulsifying Drug Delivery System (SEDDS) Using the Hybrid of Medium and Long-Chain Triglycerides


Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Drug Concentration Determination
2.3.1. Calibration Curve of in Vitro Samples
2.3.2. Calibration Curve of Mouse Plasma Samples
2.4. Formulation Optimization
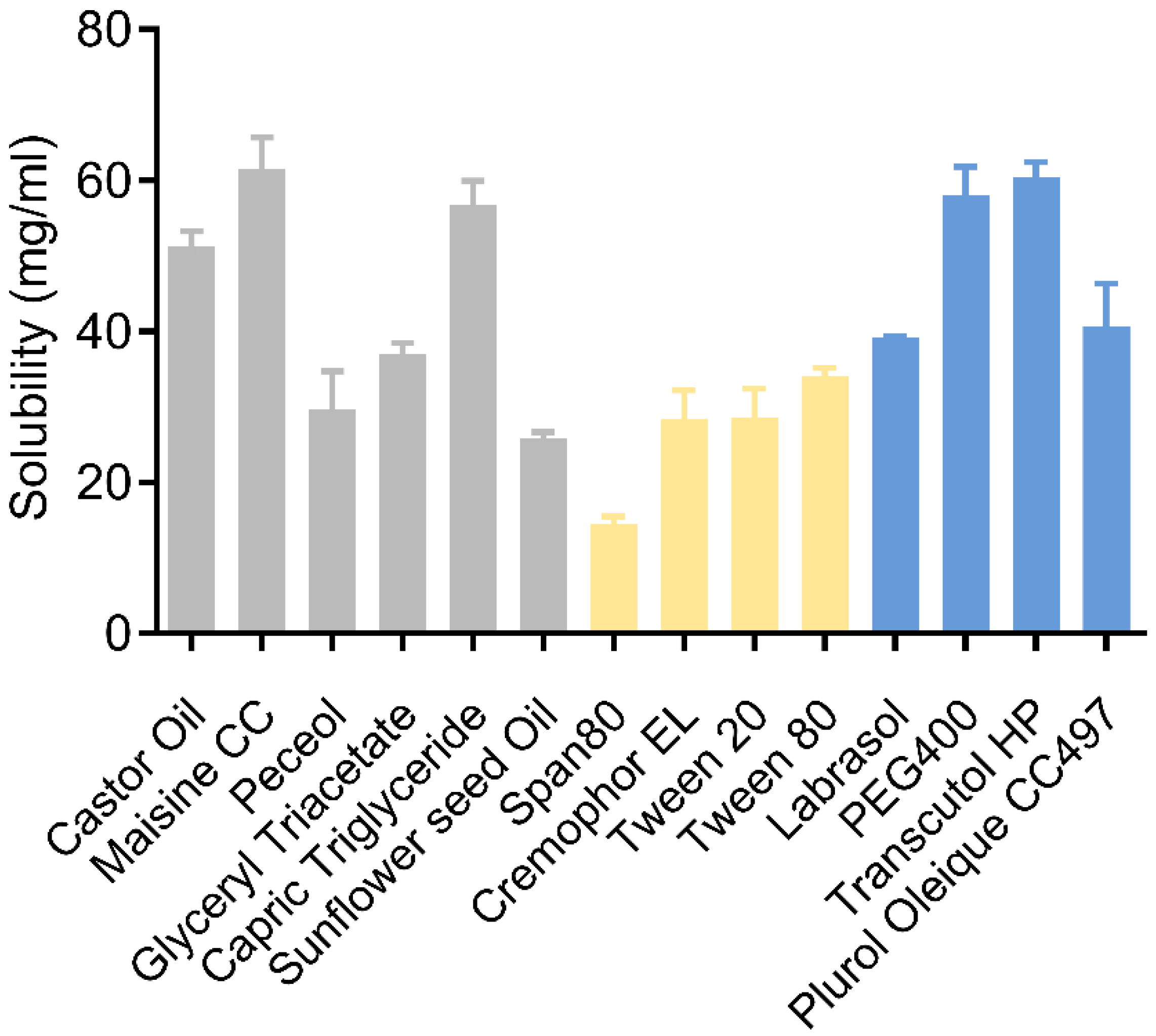
2.4.1. Determination of Drug Solubility in Excipients
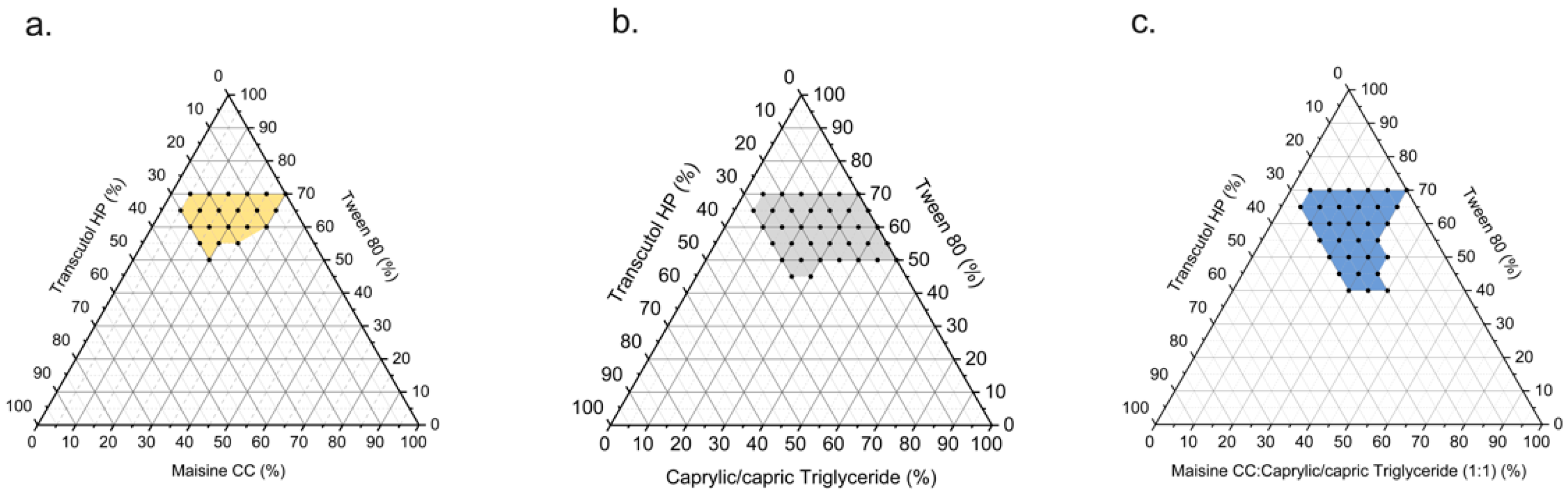
2.4.2. Pseudo-Ternary Phase Diagram Construction
2.5. Characterization
2.5.1. Formulation Preparation
2.5.2. Droplet Size Analysis and Zeta Potential Measurement
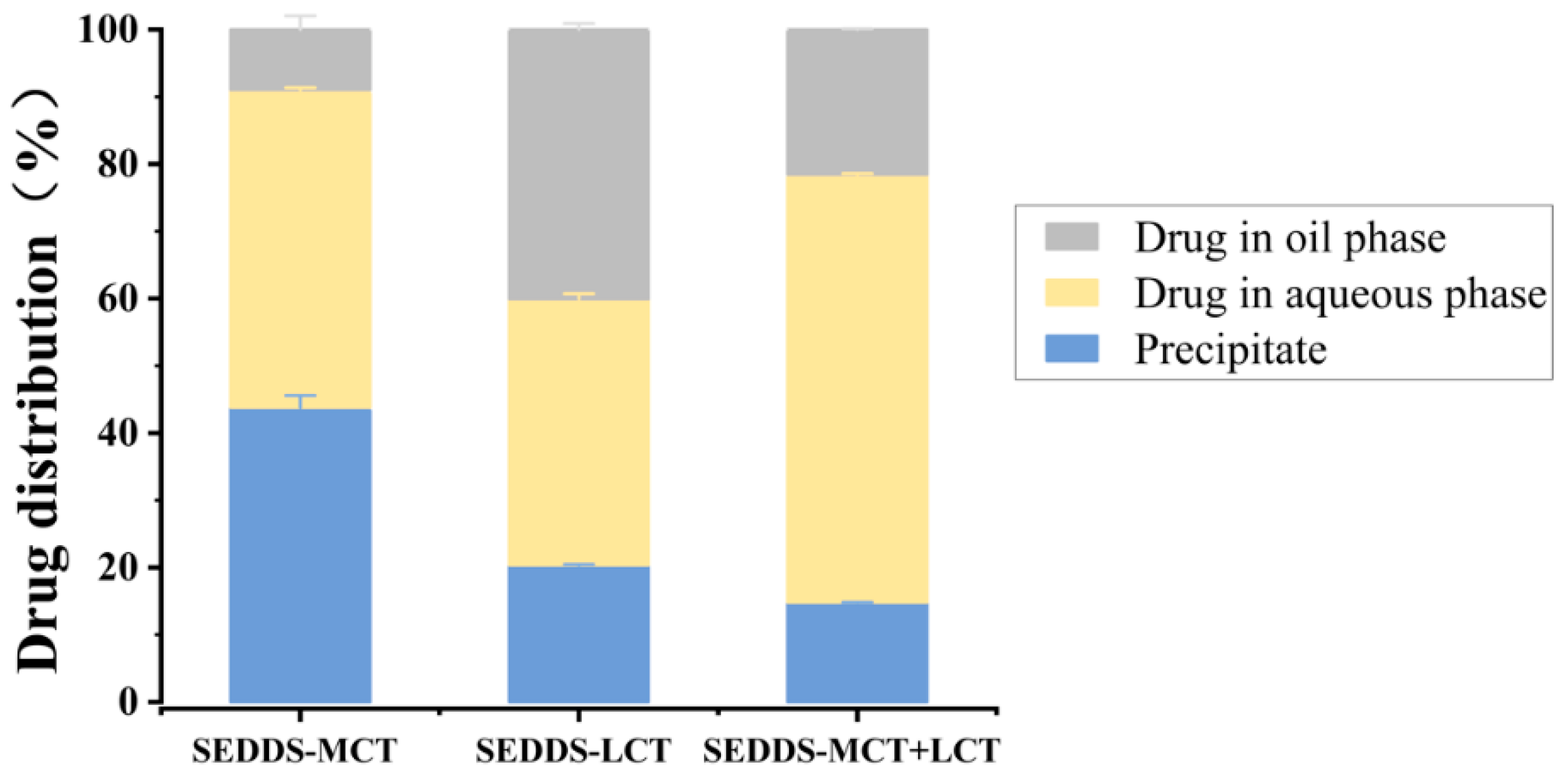
2.5.3. In Vitro Lipolysis
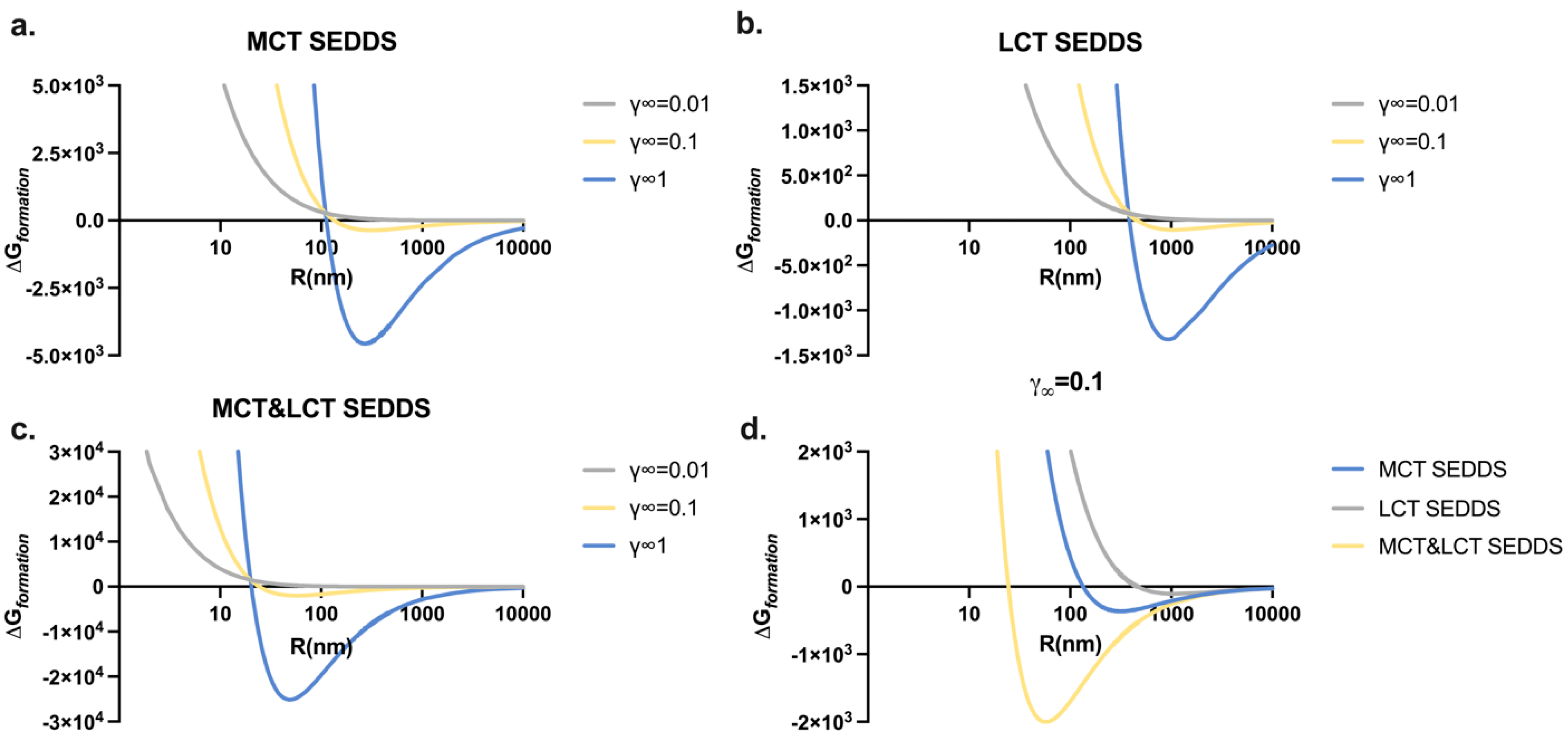
2.6. Thermodynamic Models of SEDDS
2.7. Caco-2 Cell Lines Culture and Permeability Studies
2.8. Drug Absorption Approach Study
2.8.1. Pharmacokinetic Study in ICR Mice
2.8.2. Chylomicron Blocking Model Study
2.9. Statistical Analysis
3. Results and Discussion
3.1. Solubility Study
3.2. Pseudo-Ternary Phase Diagram Construction
3.3. Droplet Size and Zeta Potential Analysis
3.4. Particle Radius Influence on the Thermodynamic Profiles of SEDDSs
3.5. In Vitro Lipolysis Study
3.6. Permeability of Progesterone
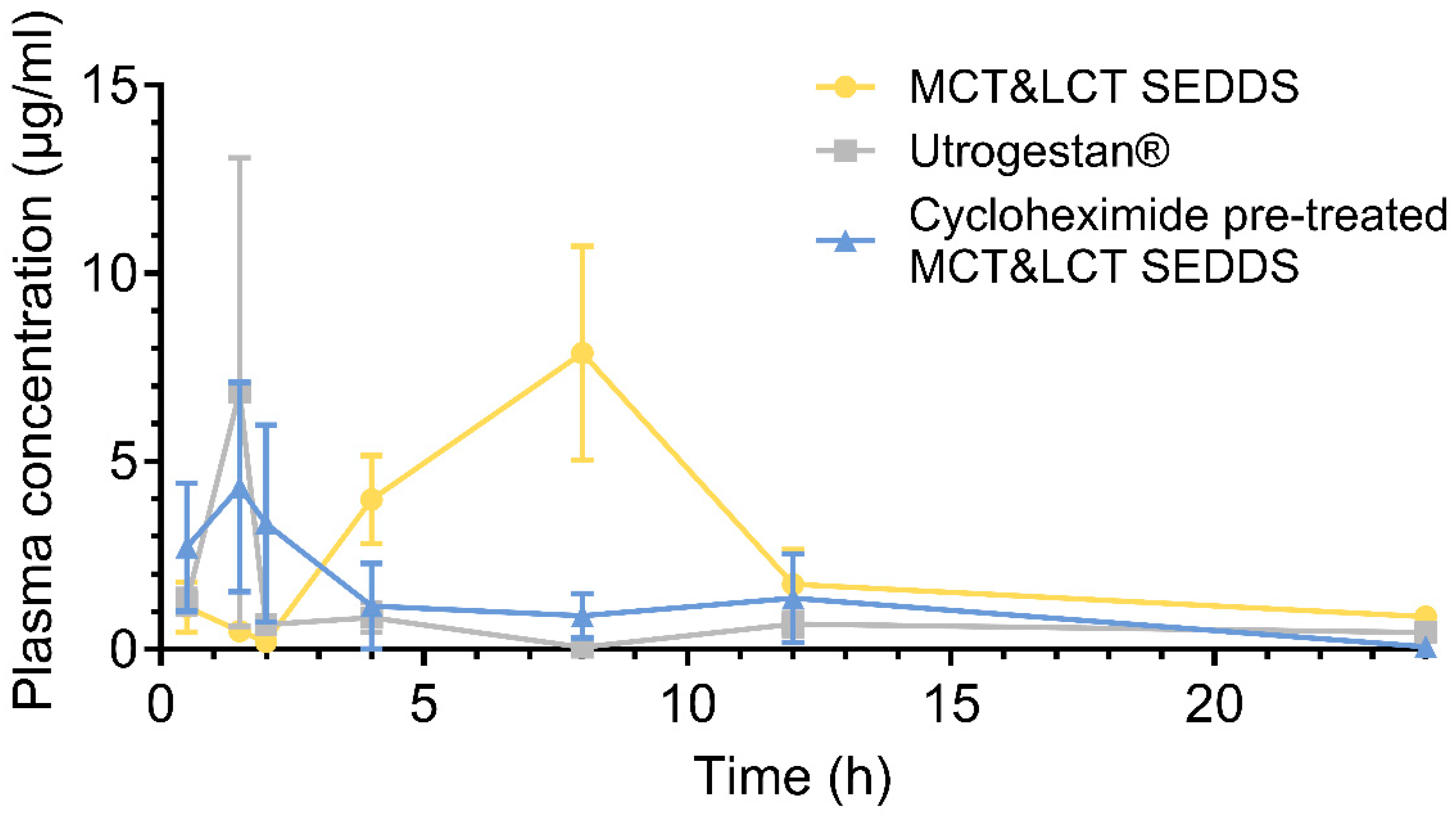
3.7. Pharmacokinetics Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FDA | Food and Drug Administration |
| HPLC | High-performance liquid chromatography |
| LCT | Long-chain length triglycerides |
| MCT | Medium-chain length triglycerides |
| SEDDS | Self-emulsifying drug delivery system |
| SOR | Surfactant-to-oil ratio |
References
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.A.; Williams, J.A. Origin and Evolution of High Throughput Screening. Br. J. Pharmacol. 2007, 152, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation Design for Poorly Water-Soluble Drugs Based on Biopharmaceutics Classification System: Basic Approaches and Practical Applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Liu, Y.; Li, X.; Yang, P.; He, W. Solubilization Techniques Used for Poorly Water-Soluble Drugs. Acta Pharm. Sin. B 2024, 14, 4683–4716. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- O’Driscoll, C.M. Lipid-Based Formulations for Intestinal Lymphatic Delivery. Eur. J. Pharm. Sci. 2002, 15, 405–415. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J.H. Lipid-Based Delivery Systems and Intestinal Lymphatic Drug Transport: A Mechanistic Update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef]
- Čerpnjak, K.; Zvonar, A.; Gašperlin, M.; Vrečer, F. Lipid-Based Systems as a Promising Approach for Enhancing the Bioavailability of Poorly Water-Soluble Drugs. Acta Pharm. 2013, 63, 427–445. [Google Scholar] [CrossRef]
- Krstić, M.; Medarević, Đ.; Đuriš, J.; Ibrić, S. Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) and Self-Microemulsifying Drug Delivery Systems (SMEDDS) as Lipid Nanocarriers for Improving Dissolution Rate and Bioavailability of Poorly Soluble Drugs; William Andrew Publishing: New York, NY, USA, 2018; ISBN 9780128136874. [Google Scholar]
- Sheth, T.; Seshadri, S.; Prileszky, T.; Helgeson, M.E. Multiple Nanoemulsions. Nat. Rev. Mater. 2020, 5, 214–228. [Google Scholar] [CrossRef]
- Nikolaev, B.; Yakovleva, L.; Fedorov, V.; Li, H.; Gao, H.; Shevtsov, M. Nano- and Microemulsions in Biomedicine: From Theory to Practice. Pharmaceutics 2023, 15, 1989. [Google Scholar] [CrossRef]
- Mariyate, J.; Bera, A. A Critical Review on Selection of Microemulsions or Nanoemulsions for Enhanced Oil Recovery. J. Mol. Liq. 2022, 353, 118791. [Google Scholar] [CrossRef]
- Griesser, J.; Hetényi, G.; Kadas, H.; Demarne, F.; Jannin, V.; Bernkop-Schnürch, A. Self-Emulsifying Peptide Drug Delivery Systems: How to Make Them Highly Mucus Permeating. Int. J. Pharm. 2018, 538, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Du, T.; Bai, Y.; Yang, T.; Zuo, J. Effects of Surfactant Molecular Structure on the Stability of Water in Oil Emulsion. J. Pet. Sci. Eng. 2021, 196, 107695. [Google Scholar] [CrossRef]
- Zembyla, M.; Murray, B.S.; Sarkar, A. Water-in-Oil Emulsions Stabilized by Surfactants, Biopolymers and/or Particles: A Review. Trends Food Sci. Technol. 2020, 104, 49–59. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, D.; Hu, J. Effect of Surfactant Molecular Structure on Emulsion Stability Investigated by Interfacial Dilatational Rheology. Polymers 2021, 13, 1127. [Google Scholar] [CrossRef]
- Salawi, A. Self-Emulsifying Drug Delivery Systems: A Novel Approach to Deliver Drugs. Drug Deliv. 2022, 29, 1811–1823. [Google Scholar] [CrossRef]
- Handa, M.; Ujjwal, R.R.; Vasdev, N.; Flora, S.J.S.; Shukla, R. Optimization of Surfactant- and Cosurfactant-Aided Pine Oil Nanoemulsions by Isothermal Low-Energy Methods for Anticholinesterase Activity. ACS Omega 2021, 6, 559–568. [Google Scholar] [CrossRef]
- Li, P.; Huang, Y.; Marshall, M.; Brooks, A.; Chin, M.; Li, J.; Wang, Y.; No, D.S.; Fang, Y.; Abbaspourrad, A. Co-Surfactant Roles of Amino Acids at Oil-Water Interface: Application in Low-PH Emulsions to Regulate Physical and Oxidative Stabilities. Food Chem. 2025, 479, 143775. [Google Scholar] [CrossRef]
- Reham, S.S.; Masjuki, H.H.; Kalam, M.A.; Shancita, I.; Rizwanul Fattah, I.M.; Ruhul, A.M. Study on Stability, Fuel Properties, Engine Combustion, Performance and Emission Characteristics of Biofuel Emulsion. Renew. Sustain. Energy Rev. 2015, 52, 1566–1579. [Google Scholar] [CrossRef]
- Smail, S.S.; Ghareeb, M.M.; Omer, H.K.; Al-Kinani, A.A.; Alany, R.G. Studies on Surfactants, Cosurfactants, and Oils for Prospective Use in Formulation of Ketorolac Tromethamine Ophthalmic Nanoemulsions. Pharmaceutics 2021, 13, 467. [Google Scholar] [CrossRef]
- Maher, S.; Geoghegan, C.; Brayden, D.J. Safety of Surfactant Excipients in Oral Drug Formulations. Adv. Drug Deliv. Rev. 2023, 202, 115086. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Ranjan Sinha, V.; Kaur, R. Clinical Considerations on Micro- and Nanodrug Delivery Systems; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 3, ISBN 9780128178706. [Google Scholar]
- Tian, C.; Guo, J.; Miao, Y.; Wang, H.; Ye, Q.; Guo, C.; Zhang, M.; He, Z.; Sun, J. Long Chain Triglyceride-Lipid Formulation Promotes the Oral Absorption of the Lipidic Prodrugs through Coincident Intestinal Behaviors. Eur. J. Pharm. Biopharm. 2022, 176, 122–132. [Google Scholar] [CrossRef]
- Watanabe, S.; Tsujino, S. Applications of Medium-Chain Triglycerides in Foods. Front. Nutr. 2022, 9, 802805. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, H.N.; Patel, D.P.; Patel, N.G.; Dalrymple, D.M.; Serajuddin, A.T.M. Effect of Difference in Fatty Acid Chain Lengths of Medium-Chain Lipids on Lipid/Surfactant/Water Phase Diagrams and Drug Solubility. J. Excip. Food Chem. 2011, 2, 73–88. [Google Scholar]
- Hamdy, A.; El-Badry, M.; Fathy, M.; El-Sayed, A.M. Impact of Oil Type on the Development and Oral Bioavailability of Self-Nanoemulsifying Drug Delivery Systems Containing Simvastatin. Sci. Rep. 2024, 14, 22584. [Google Scholar] [CrossRef] [PubMed]
- Nardin, I.; Köllner, S. Successful Development of Oral SEDDS: Screening of Excipients from the Industrial Point of View. Adv. Drug Deliv. Rev. 2019, 142, 128–140. [Google Scholar] [CrossRef]
- Prasad, D.; Chauhan, H.; Atef, E. Studying the Effect of Lipid Chain Length on the Precipitation of a Poorly Water Soluble Drug from Self-Emulsifying Drug Delivery System on Dispersion into Aqueous Medium. J. Pharm. Pharmacol. 2013, 65, 1134–1144. [Google Scholar] [CrossRef]
- Caliph, S.M.; Charman, W.N.; Porter, C.J.H. Effect of Short-, Medium-, and Long-Chain Fatty Acid-Based Vehicles on the Absolute Oral Bioavailability and Intestinal Lymphatic Transport of Halofantrine and Assessment of Mass Balance in Lymph-Cannulated and Non-Cannulated Rats. J Pharm. Sci. 2000, 89, 1073–1084. [Google Scholar] [CrossRef]
- Alayoubi, A.; Aqueel, M.S.; Cruz, C.N.; Ashraf, M.; Zidan, A.S. Application of in Vitro Lipolysis for the Development of Oral Self-Emulsified Delivery System of Nimodipine. Int. J. Pharm. 2018, 553, 441–453. [Google Scholar] [CrossRef]
- Hunter, R.J.; White, L.R.; Chan, D.Y. Foundations of Colloid Science: Volume II; Springer Nature: Cham, Switzerland, 1989. [Google Scholar]
- McClements, D.J. Nanoemulsions versus Microemulsions: Terminology, Differences, and Similarities. Soft Matter 2012, 8, 1719–1729. [Google Scholar] [CrossRef]
- Lee, C.O. Configurational Entropy and Instability of Tachyonic Braneworld. Phys. Lett. Sect. B Nucl. Elem. Part. High-Energy Phys. 2020, 800, 135030. [Google Scholar] [CrossRef]
- Hong, I.K.; Kim, S.I.; Lee, S.B. Effects of HLB Value on Oil-in-Water Emulsions: Droplet Size, Rheological Behavior, Zeta-Potential, and Creaming Index. J. Ind. Eng. Chem. 2018, 67, 123–131. [Google Scholar] [CrossRef]
- Chatterjee, B.; Hamed Almurisi, S.; Ahmed Mahdi Dukhan, A.; Mandal, U.K.; Sengupta, P. Controversies with Self-Emulsifying Drug Delivery System from Pharmacokinetic Point of View. Drug Deliv. 2016, 23, 3639–3652. [Google Scholar] [CrossRef]
- Agubata, C.O.; Nzekwe, I.T.; Obitte, N.C.; Ugwu, C.E.; Attama, A.A.; Onunkwo, G.C. Effect of Oil, Surfactant and Co-Surfactant Concentrations on the Phase Behavior, Physicochemical Properties and Drug Release from Self-Emulsifying Drug Delivery Systems. J. Drug Discov. Dev. Deliv. 2014, 1, 7. [Google Scholar]
- Pinto, I.; Buss, A. ζ Potential as a Measure of Asphalt Emulsion Stability. Energy Fuels 2020, 34, 2143–2151. [Google Scholar] [CrossRef]
- Niu, H.; Wang, W.; Dou, Z.; Chen, X.; Chen, X.; Chen, H.; Fu, X. Multiscale Combined Techniques for Evaluating Emulsion Stability: A Critical Review. Adv. Colloid Interface Sci. 2023, 311, 102813. [Google Scholar] [CrossRef]
- Dong, W.; Ye, J.; Zhou, J.; Wang, W.; Wang, H.; Zheng, X.; Yang, Y.; Xia, X.; Liu, Y. Comparative Study of Mucoadhesive and Mucus-Penetrative Nanoparticles Based on Phospholipid Complex to Overcome the Mucus Barrier for Inhaled Delivery of Baicalein. Acta Pharm. Sin. B 2020, 10, 1576–1585. [Google Scholar] [CrossRef]
- Patel, R.P.; Cristofoletti, R.; Wu, F.; Shoyaib, A.A.; Polli, J.E. In Vitro Lipolysis Model to Predict Food Effect of Poorly Water-Soluble Drugs Itraconazole, Rivaroxaban, and Ritonavir. J. Pharm. Sci. 2024, 113, 2361–2373. [Google Scholar] [CrossRef]
- Porat, D.; Dahan, A. Active Intestinal Drug Absorption and the Solubility-Permeability Interplay. Int. J. Pharm. 2018, 537, 84–93. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M. The Solubility-Permeability Interplay and Its Implications in Formulation Design and Development for Poorly Soluble Drugs. AAPS J. 2012, 14, 244–251. [Google Scholar] [CrossRef]
- Beig, A.; Fine-Shamir, N.; Porat, D.; Lindley, D.; Miller, J.M.; Dahan, A. Concomitant Solubility-Permeability Increase: Vitamin E TPGS vs. Amorphous Solid Dispersion as Oral Delivery Systems for Etoposide. Eur. J. Pharm. Biopharm. 2017, 121, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Van Breemen, R.B.; Li, Y. Caco-2 Cell Permeability Assays to Measure Drug Absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Fedi, A.; Vitale, C.; Ponschin, G.; Ayehunie, S.; Fato, M.; Scaglione, S. In Vitro Models Replicating the Human Intestinal Epithelium for Absorption and Metabolism Studies: A Systematic Review. J. Control. Release 2021, 335, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Escalera, S.; Wellejus, A. Evaluation of Caco-2 and Human Intestinal Epithelial Cells as in Vitro Models of Colonic and Small Intestinal Integrity. Biochem. Biophys. Rep. 2022, 31, 101314. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/Nda/2020/201110Orig1s000SumR.Pdf (accessed on 26 April 2025).
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/Label/2007/017362s104lbl.Pdf (accessed on 26 April 2025).
- Sahu, R.K.; Khan, J. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs. Adv. Chall. Pharm. Technol. Mater. Process Dev. Drug Deliv. Strateg. 2021, 2013, 229–242. [Google Scholar] [CrossRef]
- Walkery, A.; Leader, L.D.; Cooke, E.; Vandenberg, A. Review of Allopregnanolone Agonist Therapy for the Treatment of Depressive Disorders. Drug Des. Devel. Ther. 2021, 15, 3017–3026. [Google Scholar] [CrossRef]
- Ryšánek, P.; Grus, T.; Lukáč, P.; Kozlík, P.; Křížek, T.; Pozniak, J.; Roušarová, J.; Královičová, J.; Kutinová Canová, N.; Boleslavská, T.; et al. Validity of Cycloheximide Chylomicron Flow Blocking Method for the Evaluation of Lymphatic Transport of Drugs. Br. J. Pharmacol. 2021, 178, 4663–4674. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | MCT-SEDDS | LCT-SEDDS | MCT&LCT-SEDDS |
|---|---|---|---|
| Progesterone (g) | 1.0 | 1.0 | 1.0 |
| Maisine CC (g) | 0 | 10.0 | 5.0 |
| Capric Triglyceride (g) | 10.0 | 0 | 5.0 |
| Tween-80 (g) | 27.5 | 27.5 | 27.5 |
| Transcutol HP (g) | 12.5 | 12.5 | 12.5 |
| Composition (Weight Fractions %) | Formulation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | |
| Maisine CC | 20 | 18 | 16 | 14 | 12 | 10 | 8 | 6 | 4 | 2 | 0 |
| Capric Triglyceride | 0 | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 | 18 | 20 |
| Tween-80 | 55 | 55 | 55 | 55 | 55 | 55 | 55 | 55 | 55 | 55 | 55 |
| Transcutol HP | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 |
| MCT/LCT ratio | 0:10 | 1:9 | 2:8 | 3:7 | 4:6 | 5:5 | 6:4 | 7:3 | 8:2 | 9:1 | 10:0 |
| Formulation | Droplet Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| F1 (LCT SEDDS) | 371.60 ± 6.90 ** | 0.29 ± 0.015 | +4.35 ± 0.33 |
| F6 (MCT&LCT SEDDS) | 21.23 ± 0.30 | 0.37 ± 0.016 | −7.28 ± 0.73 |
| F11 (MCT SEDDS) | 113.50 ± 0.34 ** | 0.28 ± 0.007 | −5.49 ± 0.15 |
| Pharmacokinetics Parameters | Utrogestan® | MCT&LCT-SEDDS | Cycloheximide+ MCT&LCT-SEDDS |
|---|---|---|---|
| 8.14 ± 14.60 | 8.58 ± 6.34 | 9.19 ± 6.26 | |
| 1.33 ± 1.37 | 6.08 ± 3.17 ** | 3.58 ± 3.46 | |
| 6.69 ± 5.37 | 6.84 ± 5.08 | 3.72 ± 2.84 | |
| 17.34 ± 8.58 | 66.33 ± 37.22 * | 23.72 ± 13.61 # | |
| 21.71 ± 13.58 | 74.89 ± 35.12 ** | 26.56 ± 15.43 # | |
| 9.04 ± 5.02 | 9.16 ± 1.78 | 5.00 ± 2.47 ## | |
| 1.27 ± 0.71 | 0.34 ± 0.19 * | 1.06 ± 0.71 # | |
| Relative (compare to Utrogestan®) | - | 382.52% | 136.79% |
| Relative (compare to MCT&LCT-SEDDS) | - | - | 35.76% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, K.; Lin, Y.; Zhao, B.; Liu, X. Droplet Size Reduction of Self-Emulsifying Drug Delivery System (SEDDS) Using the Hybrid of Medium and Long-Chain Triglycerides. Pharmaceutics 2025, 17, 822. https://doi.org/10.3390/pharmaceutics17070822
Qian K, Lin Y, Zhao B, Liu X. Droplet Size Reduction of Self-Emulsifying Drug Delivery System (SEDDS) Using the Hybrid of Medium and Long-Chain Triglycerides. Pharmaceutics. 2025; 17(7):822. https://doi.org/10.3390/pharmaceutics17070822
Chicago/Turabian StyleQian, Kaijie, Yuanyuan Lin, Bingxiang Zhao, and Xiangrui Liu. 2025. "Droplet Size Reduction of Self-Emulsifying Drug Delivery System (SEDDS) Using the Hybrid of Medium and Long-Chain Triglycerides" Pharmaceutics 17, no. 7: 822. https://doi.org/10.3390/pharmaceutics17070822
APA StyleQian, K., Lin, Y., Zhao, B., & Liu, X. (2025). Droplet Size Reduction of Self-Emulsifying Drug Delivery System (SEDDS) Using the Hybrid of Medium and Long-Chain Triglycerides. Pharmaceutics, 17(7), 822. https://doi.org/10.3390/pharmaceutics17070822