
Solute–Vehicle–Skin Interactions and Their Contribution to Pharmacokinetics of Skin Delivery

 , ,
, ,  and
and 
Abstract

1. Introduction
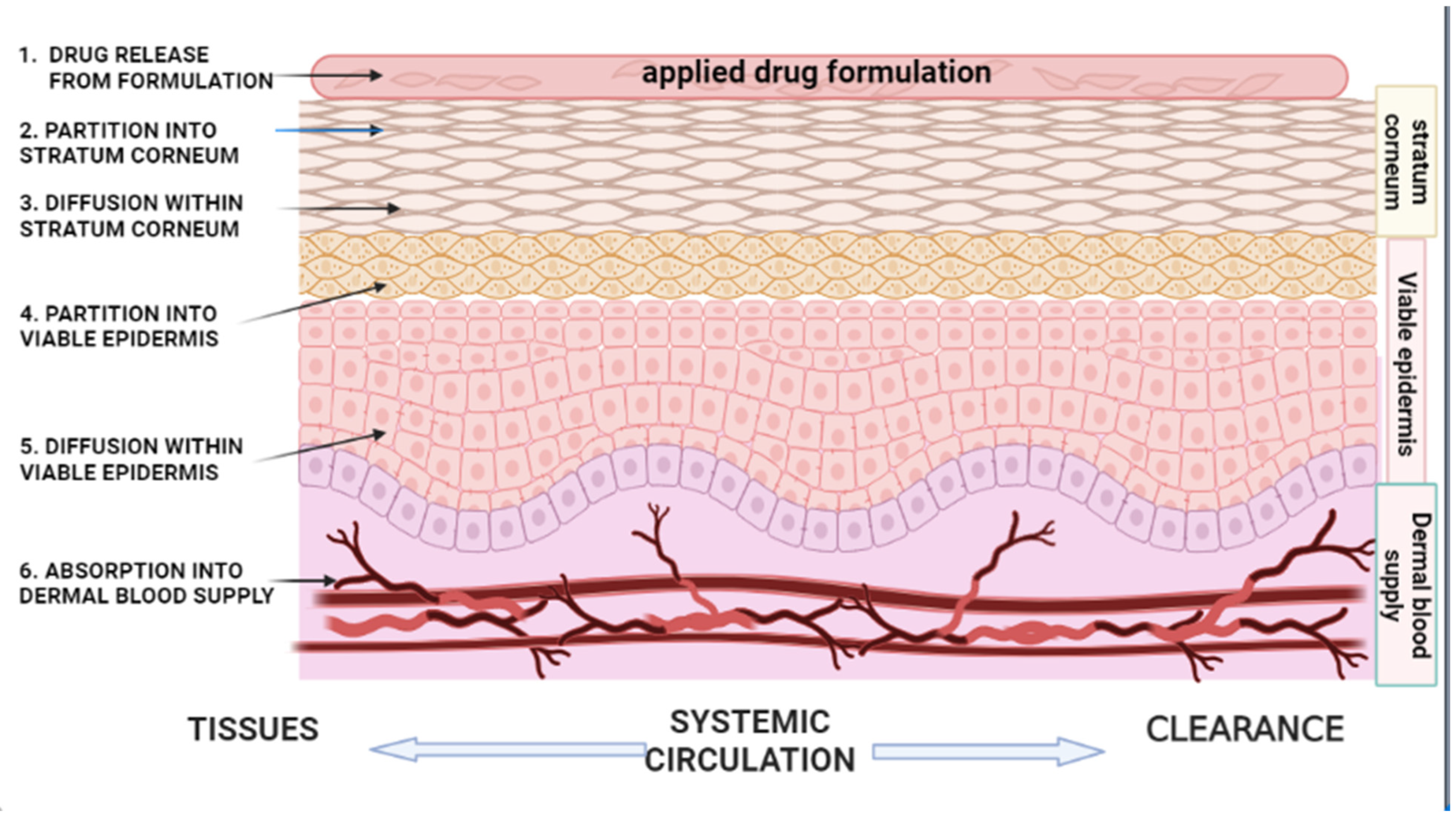
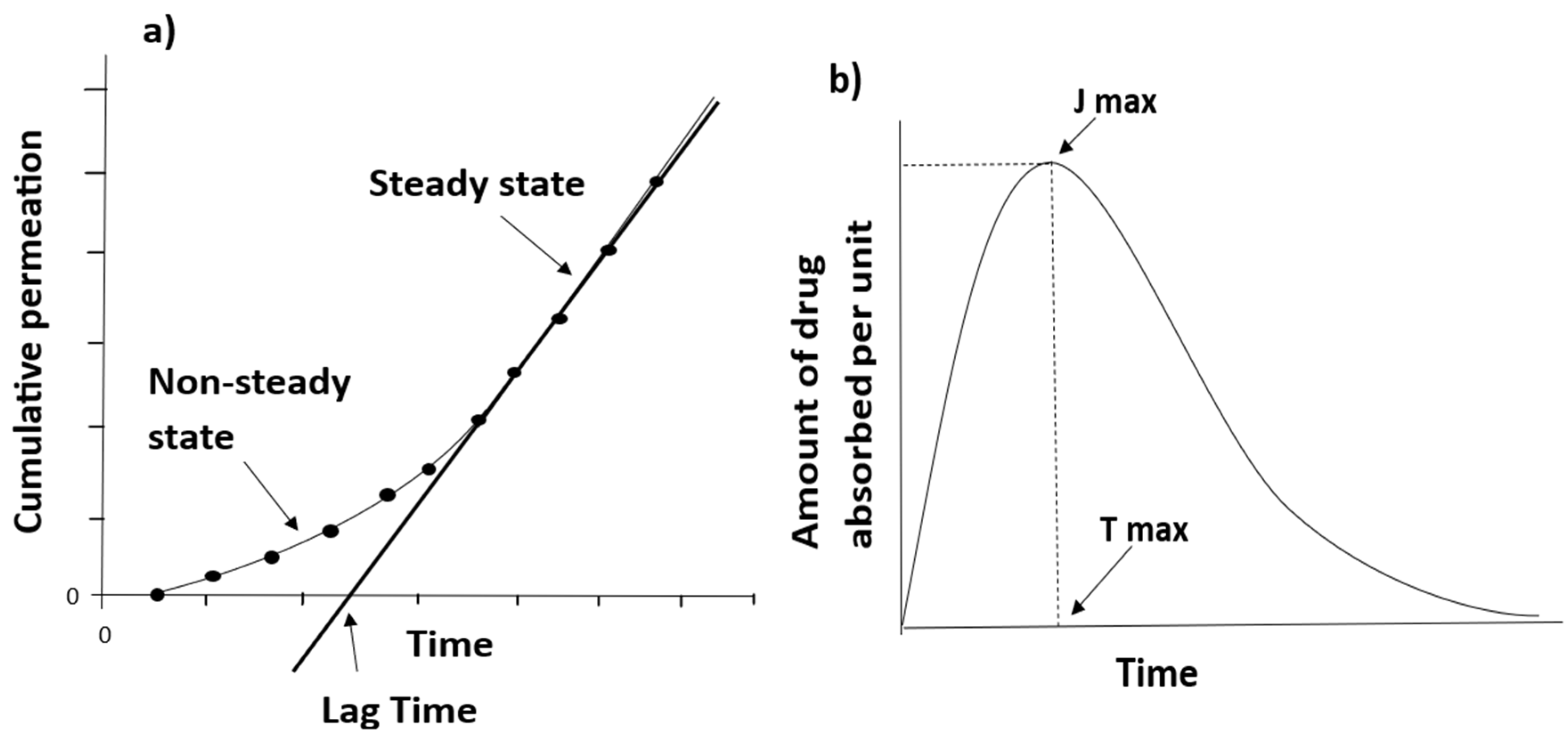
2. Kinetics of Percutaneous Absorption

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Title | Published Journal and Year | General Aims | Highlights | References |
|---|---|---|---|---|---|
| Roberts et al. | Topical drug delivery: history, percutaneous absorption, and product development | Advanced Drug Delivery Reviews, 2021 | Describes the drug delivery of topical products from the perspective of their development over time, and how an active pharmaceutical ingredient (API) gets to its target site | Quantitative structure permeability relationships (QSPR) Molecular dynamics simulations Dermal physiologically based pharmacokinetics (PBPK) Topical product delivery behavior under ‘in use’ conditions and its in vivo response | [3] |
| Supe S, Takudage P. | Methods for evaluating penetration of drug into the skin: a review | Skin Research and Technology, 2021 | Understand physiochemical characteristics impacting skin penetration of drug, as well as models employed for human skin permeation studies, and their advantages and disadvantages | Dermato-pharmacokinetics of different transdermal formulations Roles of different skin parts in percutaneous absorption and drug penetration Importance of evaluation methods and models in developing topical pharmaceutical formulations | [29] |
| Alkilani et al. | Beneath the skin: a review of current trends and future prospects of transdermal drug delivery systems | Pharmaceutics, 2022 | Discussion on recent trends in transdermal drug delivery, and their advantages and disadvantages | Methods for improving drug permeability across the skin Hurdles restricting the application of transdermal drug delivery and future prospect | [30] |
| Feschuk et al. | Regional variation in percutaneous absorption in in vitro human models: a systematic review | Journal of Toxicology and Environmental Health, 2022 | Outlines regional variation in percutaneous penetration in in vitro human models | Factors influence percutaneous penetrationImportance of ranking the susceptibility of different anatomical regions in:
| [31] |
| Jin et al. | Metamorphosis of topical semisolid products—understanding the role of rheological properties in drug permeation under the “in use” condition | Pharmaceutics, 2023 | Investigates metamorphosis of topical semisolid products—role of rheological properties in drug permeation under the “in use” condition | Effect of metamorphosis on rheological change during volatile solvent evaporation and on the permeability of API of topical semisolid formulations Role of metamorphotic events in understanding in vitro permeation profile | [32] |
| Chedik et al. | An update of skin permeability data based on a systematic review of recent research | Sci. Data, 2024 | Update available skin permeability data by compiling recently published research | Inclusion and exclusion criteria selected to build the harmonized and reusable dataset of skin permeability Influence of different experimental parameters | [33] |
| Lee et al. | Advancements in skin-mediated drug delivery: mechanisms, techniques, and applications | Advanced Healthcare Materials, 2024 | Provide an overview of skin-mediated drug delivery systems and techniques and mechanisms applied to enhance drug permeation across the skin | Skin-mediated drug delivery methods Mechanisms and techniques associated with skin-mediated drug delivery methods Recent advancements and limitations in the application of skin-mediated drug delivery | [34] |
2.1. Partition from the Vehicle/Vehicle Release
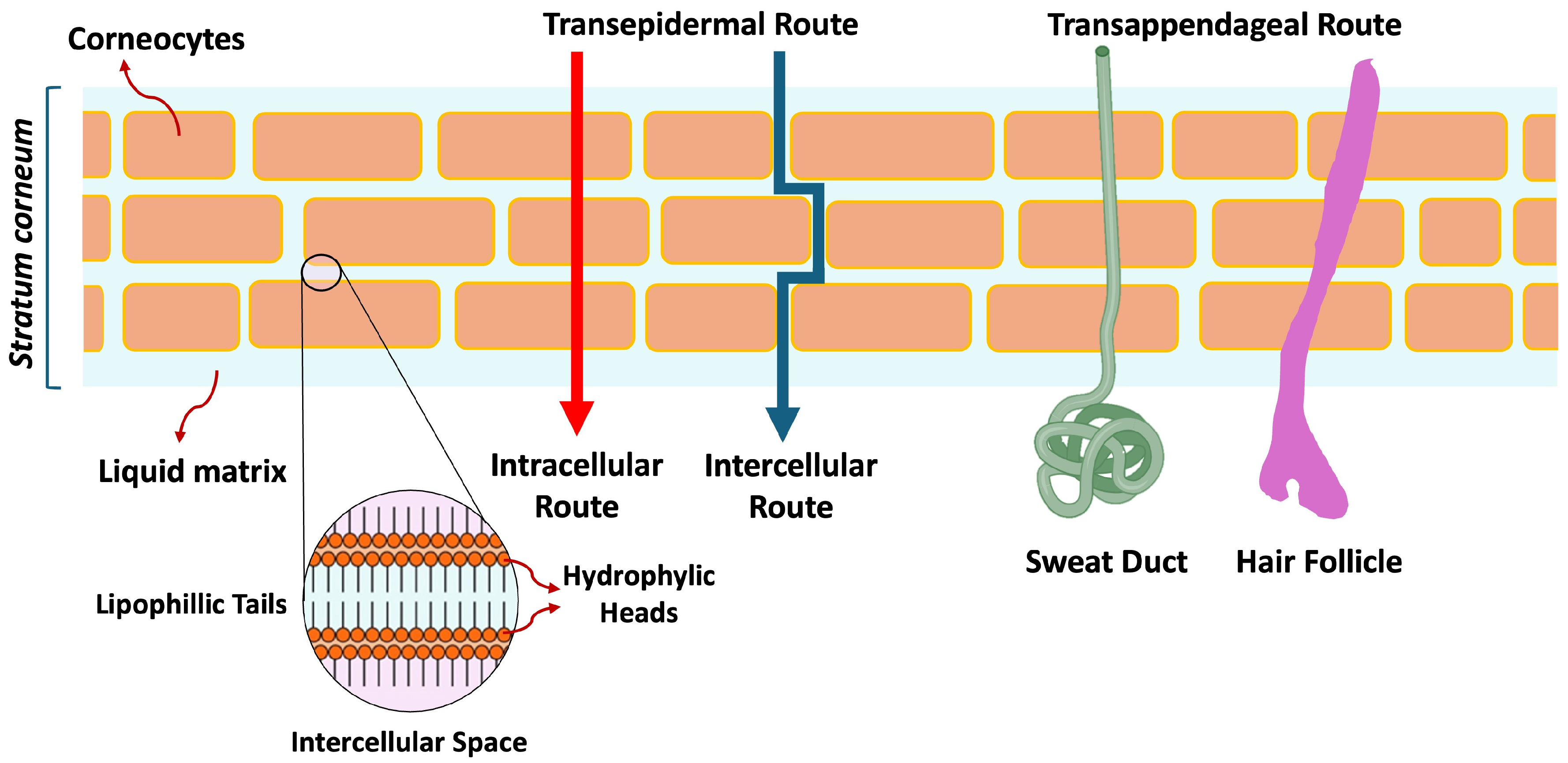
2.2. Partition into the Stratum Corneum (SC)
| Drug Name | MW(Da) | MP (°C) | Log P | Saq (mg/mL) at 25 °C | Hd | Ha |
|---|---|---|---|---|---|---|
| Acyclovir | 225.2 | 255 | −1.56 | 1.62, 2.50 (37 °C) | 3 | 5 |
| Adapalene | 412.5 | 319–322 | 8.60 | 0.000004 | 1 | 3 |
| Alclometasone dipropionate | 520.2 | 212–216 | 3.94 | 0.14 | 1 | 7 |
| Amcinonide | 502.6 | 250–252 | 2.30 | 0.0077 | 1 | 8 |
| Aminolevulinic acid hydrochloride | 167.6 | 144–151 | −2.80 | 173 | 3 | 4 |
| Amlexanox | 298.3 | 300 | 4.10 | 0.15 | 2 | 6 |
| Ammonium lactate | 107.1 | 91–94 | −0.59 | 866 | 2 | 3 |
| Amphotericin B | 924.1 | 170 | 0.80 | 0.082 | 12 | 18 |
| Avobenzone | 310.4 | 84 | 4.51 | 0.0022 | 0 | 3 |
| Azelaic acid | 188.2 | 107 | 1.57 | 2.40 | 2 | 4 |
| Bacitracin zinc | 1422.7 | 250 | −2.90 | 0.025 | 15 | 21 |
| Benzoyl peroxide | 242.2 | 103–106 | 3.46 | 0.0091 | 0 | 4 |
| Benzyl alcohol | 108.1 | 205 | 1.10 | 42.90 | 1 | 1 |
| Benzyl benzoate | 212.2 | 21, liquid | 3.97 | 0.025 | 0 | 2 |
| Betamethasone dipropionate | 504.2 | 178 | 4.07 | 0.0046 | 1 | 5 |
| Betamethasone valerate | 476.6 | 184 | 3.60 | 0.0067 | 2 | 7 |
| Bexarotene | 348.5 | 230–231 | 6.90 | 0.00015 | 1 | 2 |
| Bimatoprost | 415.6 | 63–67 | 3.20 | 0.019 | 4 | 4 |
| Brimonidine tartrate | 442.2 | 207–208 | 1.27 | 0.15 | 6 | 9 |
| Butenafine hydrochloride | 353.9 | 208–210 | 5.67 | 0.00008 | 0 | 1 |
| Calcipotriene | 412.6 | 166–168 | 4.63 | 0.014 | 3 | 3 |
| Capsaicin | 305.4 | 65 | 3.04 | 0.029 | 2 | 3 |
| Chlorhexidine gluconate | 897.8 | Liquid at 25 °C | 2.71 | 0.026 | 18 | 16 |
| Ciclopirox | 207.3 | 144 | 2.30 | 1.41 | 1 | 2 |
| Clindamycin phosphate | 504.1 | 114 | 0.93 | 3.12 | 5 | 10 |
| Clobetasol propionate | 466.2 | 196 | 3.50 | 0.0039 | 1 | 6 |
| Clocortolone pivalate | 495.0 | 231–233 | 4.36 | 0.0010 | 1 | 6 |
| Clotrimazole | 344.1 | 148 | 0.50 | 0.00049 | 0 | 1 |
| Crisaborole | 251.1 | 129–135 | 3.24 | 0.023 | 1 | 4 |
| Crotamiton | 203.3 | Liquid at 25 °C | 2.16 | 0.018 | 0 | 1 |
| Dapsone | 248.3 | 175–177 | 0.97 | 0.28, 0.38 (37 °C) | 2 | 4 |
| Delgocitinib | 310.4 | N/A | 1.29 | 0.552 | 1 | 5 |
| Desonide | 416.5 | 257–260 | 1.40 | 0.059 | 2 | 6 |
| Desoximetasone | 376.2 | 217 | 2.35 | 0.042 | 2 | 5 |
| Diclofenac sodium | 316.9 | 284 | 0.70 | 0.0048 | 1 | 3 |
| Diflorasone diacetate | 494.5 | 221–223 | 2.10 | 0.085 | 1 | 9 |
| Docosanol | 326.6 | 65–72 | 9.00 | 0.000000075 | 1 | 1 |
| Doxepin hydrochloride | 315.8 | 187–189 | 4.29 | 0.032 | 1 | 2 |
| Dyclonine hydrochloride (Dyclopro) | 325.9 | 175–176 | 4.66 | 0.049 | 1 | 3 |
| Econazole nitrate | 443.0 | 162 | 4.67 | 0.0015 | 1 | 5 |
| Efinaconazole | 348.4 | 86–89 | 3.70 | 0.32 | 1 | 6 |
| Eflornithine hydrochloride | 218.6 | 181–184 | −2.19 | 50 | 4 | 6 |
| Erythromycin | 733.5 | 191 | 3.06 | 0.46 | 5 | 14 |
| Fluocinolone acetonide | 452.2 | 266–268 | 2.48 | 0.055 | 2 | 8 |
| Fluocinonide | 494.5 | 309 | 3.19 | 0.0047 | 1 | 9 |
| Fluorouracil | 130.0 | 280–282 | −0.89 | 11.10 (22 °C) | 2 | 3 |
| Flurandrenolide | 436.5 | 247–255 | 2.88 | 0.0011 | 2 | 7 |
| Fluticasone propionate | 500.6 | 261–273 | 3.38 | 0.011 | 1 | 9 |
| Gentamicin sulfate | 516.6 | 218–237 | −3.10 | 100 | 8 | 14 |
| Halcinonide | 455.0 | 276–277 | 3.30 | 0.011 | 1 | 6 |
| Halobetasol propionate | 484.2 | 213–215 | 3.73 | 0.022 | 1 | 5 |
| Hexachlorophene | 405.8 | 164–165 | 7.54 | 0.14 | 2 | 2 |
| Hydrocortisone | 362.2 | 220 | 1.61 | 0.32 | 3 | 5 |
| Hydrocortisone butyrate | 432.6 | 210–214 | 3.21 | 0.014 | 2 | 6 |
| Hydrocortisone valerate | 446.6 | 217–220 | 3.62 | 0.0076 | 2 | 6 |
| Hydroquinone | 110.1 | 172.3 | 0.59 | 72 | 2 | 2 |
| Imiquimod | 240.3 | 292–294 | 2.70 | 6.25 | 1 | 3 |
| Ingenol mebutate | 430.5 | 153.5 | 3.12 | 0.0043 | 3 | 6 |
| Ivermectin | 875.1 | 155 | 5.83 | 0.0040 | 3 | 14 |
| Ketoconazole | 530.1 | 146 | 4.35 | 0.00029 (20 °C) | 0 | 6 |
| Lidocaine | 234.2 | 69 | 2.44 | 4.10 (30 °C) | 1 | 2 |
| Lotilaner | 596.8 | N/A | 5.81 | N/A | 2 | 4 |
| Luliconazole | 354.3 | 149–154 | 2.59 | 0.066 | 0 | 4 |
| Mafenide acetate | 246.3 | 177 | −0.37 | 5.18 | 3 | 6 |
| Mechlorethamine hydrochloride | 192.5 | 108–110 | 0.91 | 33.40 | 1 | 1 |
| Metronidazole | 171.1 | 158–160 | −0.02 | 11 | 1 | 4 |
| Miconazole nitrate | 479.1 | 159–163 | 3.26 | 0.026 | 1 | 5 |
| Minoxidil | 209.3 | 248 | 1.24 | 2.20 | 3 | 3 |
| Mitomycin | 334.3 | 360 | −0.40 | 8.43 | 3 | 8 |
| Mometasone furoate | 521.4 | 218–220 | 3.90 | 0.011 | 1 | 6 |
| Mupirocin | 500.2 | 77–78 | 3.44 | 0.027 | 4 | 9 |
| Naftifine hydrochloride | 323.9 | 172–175 | 3.59 | 0.00023 | 1 | 1 |
| Neomycin sulfate | 712.7 | 187 | −7.80 | 50 | 15 | 23 |
| Nystatin | 926.1 | 160 | 0.50 | 0.36 | 12 | 18 |
| Octinoxate | 290.4 | Liquid at 25 °C | 5.80 | 0.00045 | 0 | 3 |
| Oxiconazole nitrate | 492.1 | 137 | 3.80 | 0.0019 | 1 | 6 |
| Oxybenzone | 228.2 | 65.5 | 3.79 | 0.0037 | 1 | 3 |
| Oxymetazoline hydrochloride | 296.2 | 181–183 | 4.87 | 0.0014 | 3 | 2 |
| Penciclovir | 253.3 | 275–277 | −1.10 | 7.45 | 4 | 5 |
| Permethrin | 391.3 | 34 | 6.50 | cis isomer-0.00020, trans isomer-0.00013 | 0 | 3 |
| Pimecrolimus | 810.4 | 135–136 | 4.40 | 0.0015 | 2 | 11 |
| Podofilox (Condylox) | 414.4 | 228 | 2.01 | 0.15 | 1 | 8 |
| Polymyxin B sulfate | 1301.6 | 217–220 | −0.74 | 0.074 | 20 | 29 |
| Prednicarbate | 488.6 | 114–116 | 2.92 | 0.0056 | 1 | 8 |
| Prilocaine | 220.2 | 37–38 | 2.11 | 0.54 | 2 | 2 |
| Retapamulin | 517.8 | 125–127 | 5.00 | 0.00039 | 1 | 6 |
| Roflumilast | 403.2 | 158 | 4.47 | 0.0062 | 1 | 6 |
| Ruxolitinib | 306.4 | 86 | 2.94 | 0.116 | 1 | 4 |
| Sertaconazole nitrate | 500.8 | 158–160 | 3.33 | 0.0064 | 1 | 6 |
| Silver sulfadiazine | 357.1 | 285 | 0.39 | 7.87 | 1 | 6 |
| Spinosad | 1287.7 | 112–123 | 4.00 | 0.0024 | 2 | 19 |
| Sulconazole nitrate | 460.8 | 130 | 3.21 | 0.0013 | 1 | 5 |
| Sulfacetamide sodium | 236.2 | 257 | −0.96 | 50 | 1 | 5 |
| Tacrolimus | 804.0 | 126 | 3.30 | 0.000018 | 3 | 12 |
| Tapinarof | 254.3 | 140–142 | 4.25 | 0.0339 | 2 | 2 |
| Tazarotene | 351.5 | 103–105 | 3.38 | 0.00075 | 0 | 4 |
| Terbinafine hydrochloride | 327.9 | 204–208 | 3.30 | 0.00074 | 1 | 1 |
| Tetracaine | 264.4 | 41–45 | 3.51 | 0.56 | 1 | 4 |
| Tretinoin | 300.2 | 180–182 | 6.30 | 0.0048 | 1 | 2 |
| Triamcinolone acetonide | 434.2 | 293 | 2.53 | 0.08 | 2 | 7 |
| Trifarotene | 459.6 | 245 | 6.12 | 0.00095 | 2 | 5 |
2.3. Diffusion Within Stratum Corneum (SC)
2.4. Partition into the Viable Epidermis
2.5. Diffusion Within the Viable Epidermis
2.6. Absorption into the Dermal Blood Supply (Systemic Circulation)
3. Solute–Vehicle–Skin Interactions and Percutaneous Absorption
3.1. Vehicle–Drug Interactions
3.2. Skin–Vehicle Interactions
3.3. Skin–Drug Interactions
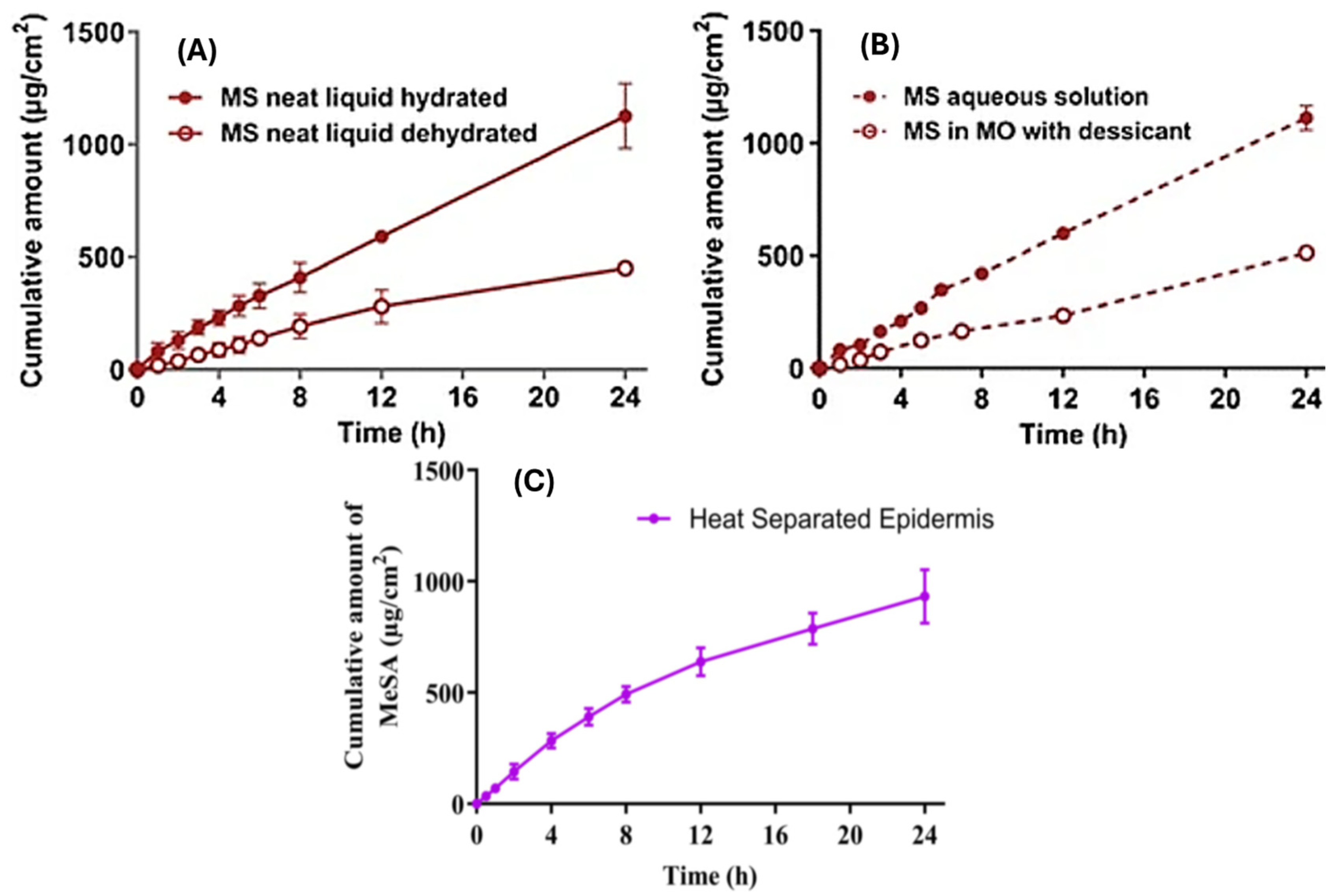
3.4. Metamorphosis of Topical Vehicles—Complex Drug–Skin–Vehicle Effects
4. Conclusions, Future Perspectives, and Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohammed, Y.; Holmes, A.; Kwok, P.C.L.; Kumeria, T.; Namjoshi, S.; Imran, M.; Matteucci, L.; Ali, M.; Tai, W.; Benson, H.A.E.; et al. Advances and future perspectives in epithelial drug delivery. Adv. Drug Deliv. Rev. 2022, 186, 114293. [Google Scholar] [CrossRef] [PubMed]
- Albayati, N.; Talluri, S.R.; Dholaria, N.; Michniak-Kohn, B. AI-Driven Innovation in Skin Kinetics for Transdermal Drug Delivery: Overcoming Barriers and Enhancing Precision. Pharmaceutics 2025, 17, 188. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Cheruvu, H.S.; Mangion, S.E.; Alinaghi, A.; Benson, H.A.E.; Mohammed, Y.; Holmes, A.; van der Hoek, J.; Pastore, M.; Grice, J.E. Topical drug delivery: History, percutaneous absorption, and product development. Adv. Drug Deliv. Rev. 2021, 177, 113929. [Google Scholar] [CrossRef] [PubMed]
- Phan, K.; Leite-Silva, V.R.; Prakash, S.; Namjoshi, S.; Panchal, B.; Roberts, M.; Mohammed, Y. Dermatological Product Sensorial Properties: Bridging Pharmaceutical and Therapeutic Equivalence. In Topical Products and Dermal Drug Delivery; Murthy, S.N., Ed.; Springer Nature: Cham, Switzerland, 2025; pp. 223–240. [Google Scholar] [CrossRef]
- Phatale, V.; Vaiphei, K.K.; Jha, S.; Patil, D.; Agrawal, M.; Alexander, A. Overcoming skin barriers through advanced transdermal drug delivery approaches. J. Control. Release 2022, 351, 361–380. [Google Scholar] [CrossRef]
- Lu, B.; Liu, T.; Wang, H.; Wu, C.; Chen, H.; Liu, Z.; Zhang, J. Ionic liquid transdermal delivery system: Progress, prospects, and challenges. J. Mol. Liq. 2022, 351, 118643. [Google Scholar] [CrossRef]
- Xiang, H.; Xu, S.; Zhang, W.; Li, Y.; Zhou, Y.; Miao, X. Skin permeation of curcumin nanocrystals: Effect of particle size, delivery vehicles, and permeation enhancer. Colloids Surf. B Biointerfaces 2023, 224, 113203. [Google Scholar] [CrossRef]
- Xiang, H.; Xu, S.; Li, J.; Pan, S.; Miao, X. Particle Size Effect of Curcumin Nanocrystals on Transdermal and Transfollicular Penetration by Hyaluronic Acid-Dissolving Microneedle Delivery. Pharmaceuticals 2022, 15, 206. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.W.; Wiemann, S.; Keck, C.M. Improved Dermal and Transdermal Delivery of Curcumin with SmartFilms and Nanocrystals. Molecules 2021, 26, 1633. [Google Scholar] [CrossRef]
- Zhang, Q.; Alinaghi, A.; Williams, D.B.; Roberts, M.S. A thermodynamic and kinetic analysis of human epidermal penetration of phenolic compounds: II. Maximum flux and solute diffusion through stratum corneum lipids. Int. J. Pharm. 2023, 631, 122522. [Google Scholar] [CrossRef]
- Tapfumaneyi, P.; Imran, M.; Alavi, S.E.; Mohammed, Y. Science of, and insights into, thermodynamic principles for dermal formulations. Drug Discov. Today 2023, 28, 103521. [Google Scholar] [CrossRef]
- Souto, E.B.; Fangueiro, J.F.; Fernandes, A.R.; Cano, A.; Sanchez-Lopez, E.; Garcia, M.L.; Severino, P.; Paganelli, M.O.; Chaud, M.V.; Silva, A.M. Physicochemical and biopharmaceutical aspects influencing skin permeation and role of SLN and NLC for skin drug delivery. Heliyon 2022, 8, e08938. [Google Scholar] [CrossRef] [PubMed]
- Neubert, R.H.H. Mechanisms of penetration and diffusion of drugs and cosmetic actives across the human Stratum corneum. Eur. J. Pharm. Biopharm. 2024, 202, 114394. [Google Scholar] [CrossRef]
- Gaikwad, S.S.; Zanje, A.L.; Somwanshi, J.D. Advancements in transdermal drug delivery: A comprehensive review of physical penetration enhancement techniques. Int. J. Pharm. 2024, 652, 123856. [Google Scholar] [CrossRef]
- Alkilani, A.Z.; McCrudden, M.T.C.; Donnelly, R.F. Transdermal Drug Delivery: Innovative Pharmaceutical Developments Based on Disruption of the Barrier Properties of the stratum corneum. Pharmaceutics 2015, 7, 438–470. [Google Scholar] [CrossRef]
- Idson, B. Percutaneous absorption. J. Pharm. Sci. 1975, 64, 901–924. [Google Scholar] [CrossRef] [PubMed]
- Riviere, J.E.; Bowman, K.F.; Monteiro-Riviere, N.A.; Dix, L.P.; Carver, M.P. The isolated perfused porcine skin flap (IPPSF): I. A novel in vitro model for percutaneous absorption and cutaneous toxicology studies. Fundam. Appl. Toxicol. 1986, 7, 444–453. [Google Scholar] [CrossRef]
- Kasting, G.B.; Miller, M.A.; Xu, L.; Yu, F.; Jaworska, J. In Vitro Human Skin Absorption of Solvent-deposited Solids: Niacinamide and Methyl Nicotinate. J. Pharm. Sci. 2022, 111, 727–733. [Google Scholar] [CrossRef]
- Tapfumaneyi, P.; Rath, S.; Bon, C.; Kanfer, I. Fitting Pharmacodynamic Data to the E(max) Model to Assess the Inherent Potency of Topical Corticosteroids. Mol. Pharm. 2022, 19, 2900–2906. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.C. Topical and transdermal drug delivery. In Aulton’s Pharmaceutics: The Design and Manufacture of Medicines, 5th ed.; Elsevier: Edinburgh, UK, 2018; pp. 715–738. [Google Scholar]
- Higuchi, T. Physical Chemical analysis of Percutaneous Absorption Process from Creams and Ointments. J. Soc. Cosmet. Chem. 1960, 11, 85–97. [Google Scholar]
- Kolimi, P.; Narala, S.; Youssef, A.A.A.; Nyavanandi, D.; Dudhipala, N. A systemic review on development of mesoporous nanoparticles as a vehicle for transdermal drug delivery. Nanotheranostics 2023, 7, 70–89. [Google Scholar] [CrossRef]
- Pulsoni, I.; Lubda, M.; Aiello, M.; Fedi, A.; Marzagalli, M.; von Hagen, J.; Scaglione, S. Comparison Between Franz Diffusion Cell and a novel Micro-physiological System for In Vitro Penetration Assay Using Different Skin Models. SLAS Technol. 2022, 27, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Magnano, G.C.; Marussi, G.; Pavoni, E.; Adami, G.; Larese Filon, F.; Crosera, M. Percutaneous metals absorption following exposure to road dust powder. Environ. Pollut. 2022, 292, 118353. [Google Scholar] [CrossRef]
- Scheuplein, R.J.; Blank, I.H. Permeability of the skin. Physiol. Rev. 1971, 51, 702–747. [Google Scholar] [CrossRef]
- Barr, M. Percutaneous absorption. J. Pharm. Sci. 1962, 51, 395–409. [Google Scholar] [CrossRef]
- Malkinson, F.D.; Ferguson, E.H. Percutaneous absorption of hydrocortisone-4-C14 in two human subjects. J. Investig. Dermatol. 1955, 25, 281–283. [Google Scholar] [CrossRef]
- Rothman, S. The Principles of Percutaneous Absorption. J. Lab. Clin. Med. 1943, 28, 1305–1321. [Google Scholar]
- Supe, S.; Takudage, P. Methods for evaluating penetration of drug into the skin: A review. Skin. Res. Technol. 2021, 27, 299–308. [Google Scholar] [CrossRef]
- Alkilani, A.Z.; Nasereddin, J.; Hamed, R.; Nimrawi, S.; Hussein, G.; Abo-Zour, H.; Donnelly, R.F. Beneath the Skin: A Review of Current Trends and Future Prospects of Transdermal Drug Delivery Systems. Pharmaceutics 2022, 14, 1152. [Google Scholar] [CrossRef] [PubMed]
- Feschuk, A.M.; Kashetsky, N.; Chiang, C.; Burli, A.; Burdick, H.; Maibach, H.I. Regional variation in percutaneous absorption in in vitro human models: A systematic review. J. Toxicol. Environ. Health B Crit. Rev. 2022, 25, 97–112. [Google Scholar] [CrossRef]
- Jin, X.; Alavi, S.E.; Shafiee, A.; Leite-Silva, V.R.; Khosrotehrani, K.; Mohammed, Y. Metamorphosis of Topical Semisolid Products-Understanding the Role of Rheological Properties in Drug Permeation under the “in Use” Condition. Pharmaceutics 2023, 15, 1707. [Google Scholar] [CrossRef]
- Chedik, L.; Baybekov, S.; Cosnier, F.; Marcou, G.; Varnek, A.; Champmartin, C. An update of skin permeability data based on a systematic review of recent research. Sci. Data 2024, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lim, S.; Kwak, S.S.; Kim, J. Advancements in Skin-Mediated Drug Delivery: Mechanisms, Techniques, and Applications. Adv. Healthc. Mater. 2024, 13, e2302375. [Google Scholar] [CrossRef] [PubMed]
- Wester, R.C.; Maibach, H.I. Percutaneous absorption of drugs. Clin. Pharmacokinet. 1992, 23, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Riegelman, S. Pharmacokinetics. Pharmacokinetic factors affecting epidermal penetration and percutaneous adsorption. Clin. Pharmacol. Ther. 1974, 16, 873–883. [Google Scholar] [CrossRef]
- Stoughton, R.B. Percutaneous absorption of drugs. Annu. Rev. Pharmacol. Toxicol. 1989, 29, 55–69. [Google Scholar] [CrossRef]
- Mckenzie, A.W.; Stoughton, R.B. Method for Comparing Percutaneous Absorption of Steroids. Arch. Dermatol. 1962, 86, 608–610. [Google Scholar] [CrossRef]
- Hollander, J.L.; Stoner, E.K.; Brown, E.M. The Use of Intra-Articular Temperature Measurement in the Evaluation of Anti-Arthritic Agents. J. Clin. Investig. 1950, 29, 822–823. [Google Scholar]
- Mckenzie, A.W. Percutaneous Absorption of Steroids. Arch. Dermatol. 1962, 86, 611–614. [Google Scholar] [CrossRef]
- Rath, S.; Zvidzayi, M.; Bon, C.; Kanfer, I. Application of E(max) model to assess the potency of topical corticosteroid products. Basic. Clin. Pharmacol. Toxicol. 2022, 131, 165–173. [Google Scholar] [CrossRef]
- Zvidzayi, M.; Rath, S.; Bon, C.; Abboo, S.; Kanfer, I. A Novel Approach to Assess the Potency of Topical Corticosteroids. Pharmaceutics 2021, 13, 1456. [Google Scholar] [CrossRef]
- High, W.A.; Prok, L.D. Dermatology Secrets E-Book: Dermatology Secrets E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Hmingthansanga, V.; Singh, N.; Banerjee, S.; Manickam, S.; Velayutham, R.; Natesan, S. Improved Topical Drug Delivery: Role of Permeation Enhancers and Advanced Approaches. Pharmaceutics 2022, 14, 2818. [Google Scholar] [CrossRef] [PubMed]
- Lunter, D.; Klang, V.; Eichner, A.; Savic, S.M.; Savic, S.; Lian, G.P.; Erdo, F. Progress in Topical and Transdermal Drug Delivery Research-Focus on Nanoformulations. Pharmaceutics 2024, 16, 817. [Google Scholar] [CrossRef] [PubMed]
- Surber, C.; Wilhelm, K.P.; Hori, M.; Maibach, H.I.; Guy, R.H. Optimization of Topical Therapy—Partitioning of Drugs into Stratum-Corneum. Pharm. Res. 1990, 7, 1320–1324. [Google Scholar] [CrossRef]
- Wang, L.M.; Chen, L.J.; Lian, G.P.; Han, L.J. Determination of partition and binding properties of solutes to stratum corneum. Int. J. Pharm. 2010, 398, 114–122. [Google Scholar] [CrossRef]
- Lboutounne, Y.; Muret, P. In vivo Skin Absorption and Skin Pharmacology. In Agache’s Measuring the Skin: Non-Invasive Investigations, Physiology, Normal Constants; Humbert, P., Fanian, F., Maibach, H.I., Agache, P., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1091–1113. [Google Scholar] [CrossRef]
- Franz, T.J. Kinetics of cutaneous drug penetration. Int. J. Dermatol. 1983, 22, 499–505. [Google Scholar] [CrossRef]
- Schafer, N.; Balwierz, R.; Biernat, P.; Ochedzan-Siodlak, W.; Lipok, J. Natural Ingredients of Transdermal Drug Delivery Systems as Permeation Enhancers of Active Substances through the. Mol. Pharm. 2023, 20, 3278–3297. [Google Scholar] [CrossRef] [PubMed]
- Wanat, K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol. Biol. Rep. 2020, 47, 3221–3231. [Google Scholar] [CrossRef]
- Raykar, P.V.; Fung, M.C.; Anderson, B.D. The role of protein and lipid domains in the uptake of solutes by human stratum corneum. Pharm. Res. 1988, 5, 140–150. [Google Scholar] [CrossRef]
- Nitsche, J.M.; Wang, T.F.; Kasting, G.B. A two-phase analysis of solute partitioning into the stratum corneum. J. Pharm. Sci. 2006, 95, 649–666. [Google Scholar] [CrossRef]
- Pastore, M.N.; Kalia, Y.N.; Horstmann, M.; Roberts, M.S. Transdermal patches: History, development and pharmacology. Br. J. Pharmacol. 2015, 172, 2179–2209. [Google Scholar] [CrossRef]
- Cross, S.E.; Anderson, C.; Roberts, M.S. Topical penetration of commercial salicylate esters and salts using human isolated skin and clinical microdialysis studies. Br. J. Clin. Pharm. 1998, 46, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Gujarathi, N.A.; Abriata, J.P.; Keservani, R.K.; Sharma, A.K. (Eds.) Topical and Transdermal Drug Delivery Systems: Applications and Future Prospects, 1st ed.; Apple Academic Press: Williston, VT, USA, 2023. [Google Scholar] [CrossRef]
- Naser, Y.A.; Tekko, I.A.; Vora, L.K.; Peng, K.; Anjani, Q.K.; Greer, B.; Elliott, C.; McCarthy, H.O.; Donnelly, R.F. Hydrogel-forming microarray patches with solid dispersion reservoirs for transdermal long-acting microdepot delivery of a hydrophobic drug. J. Control. Release 2023, 356, 416–433. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Namjoshi, S.; Benson, H.A.E.; Kumeria, T.; Mohammed, Y. Skin biomechanics: Breaking the dermal barriers with microneedles. Nano TransMed 2022, 1, e9130002. [Google Scholar] [CrossRef]
- Pires, P.C.; Mascarenhas-Melo, F.; Pedrosa, K.; Lopes, D.; Lopes, J.; Macário-Soares, A.; Peixoto, D.; Giram, P.S.; Veiga, F.; Paiva-Santos, A.C. Polymer-based biomaterials for pharmaceutical and biomedical applications: A focus on topical drug administration. Eur. Polym. J. 2023, 187, 111868. [Google Scholar] [CrossRef]
- Josiah, A.J.; Twilley, D.; Pillai, S.K.; Ray, S.S.; Lall, N. Pathogenesis of Keratinocyte Carcinomas and the Therapeutic Potential of Medicinal Plants and Phytochemicals. Molecules 2021, 26, 1979. [Google Scholar] [CrossRef]
- Carpentieri-Rodrigues, L.N.; Zanluchi, J.M.; Grebogi, I.H. Percutaneous absorption enhancers: Mechanisms and potential. Braz. Arch. Biol. Technol. 2007, 50, 949–961. [Google Scholar] [CrossRef]
- Hadgraft, J. Skin deep. Eur. J. Pharm. Biopharm. 2004, 58, 291–299. [Google Scholar] [CrossRef]
- Ramadon, D.; McCrudden, M.T.C.; Courtenay, A.J.; Donnelly, R.F. Enhancement strategies for transdermal drug delivery systems: Current trends and applications. Drug Deliv. Transl. Res. 2022, 12, 758–791. [Google Scholar] [CrossRef]
- Zhang, A.; Jung, E.C.; Zhu, H.; Zou, Y.; Hui, X.; Maibach, H. Vehicle effects on human stratum corneum absorption and skin penetration. Toxicol. Ind. Health 2017, 33, 416–425. [Google Scholar] [CrossRef]
- Dragicevic, N.; Atkinson, J.P.; Maibach, H.I. Chemical Penetration Enhancers: Classification and Mode of Action. In Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Modification of the Stratum Corneum; Dragicevic, N., Maibach, H.I., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 11–27. [Google Scholar] [CrossRef]
- Biondo, N.E.; Argenta, D.F.; Caon, T. A Comparative Analysis of Biological and Synthetic Skin Models for Drug Transport Studies. Pharm. Res. 2023, 40, 1209–1221. [Google Scholar] [CrossRef]
- Liu, M.; Wen, J.; Sharma, M. Solid Lipid Nanoparticles for Topical Drug Delivery: Mechanisms, Dosage Form Perspectives, and Translational Status. Curr. Pharm. Des. 2020, 26, 3203–3217. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Bian, Q.; Zhou, Y.; Huang, Q.; Gao, J. Hair follicle-targeting drug delivery strategies for the management of hair follicle-associated disorders. Asian J. Pharm. Sci. 2022, 17, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Christmann, R.; Ho, D.K.; Wilzopolski, J.; Lee, S.; Koch, M.; Loretz, B.; Vogt, T.; Bäumer, W.; Schaefer, U.F.; Lehr, C.M. Tofacitinib Loaded Squalenyl Nanoparticles for Targeted Follicular Delivery in Inflammatory Skin Diseases. Pharmaceutics 2020, 12, 1131. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Cavaco-Paulo, A.; Matamá, T. Mapping hair follicle-targeted delivery by particle systems: What has science accomplished so far? Int. J. Pharm. 2021, 610, 121273. [Google Scholar] [CrossRef]
- Ferreira-Nunes, R.; Cunha-Filho, M.; Gratieri, T.; Gelfuso, G.M. Follicular-targeted delivery of spironolactone provided by polymeric nanoparticles. Colloids Surf. B Biointerfaces 2021, 208, 112101. [Google Scholar] [CrossRef]
- Tolentino, S.; Pereira, M.N.; Cunha-Filho, M.; Gratieri, T.; Gelfuso, G.M. Targeted clindamycin delivery to pilosebaceous units by chitosan or hyaluronic acid nanoparticles for improved topical treatment of acne vulgaris. Carbohydr. Polym. 2021, 253, 117295. [Google Scholar] [CrossRef]
- Ban, C.; Park, J.B.; Cho, S.; Kim, H.R.; Kim, Y.J.; Choi, Y.J.; Chung, W.J.; Kweon, D.H. Reduction of focal sweating by lipid nanoparticle-delivered myricetin. Sci. Rep. 2020, 10, 13132. [Google Scholar] [CrossRef]
- Richard, C.; Cassel, S.; Blanzat, M. Vesicular systems for dermal and transdermal drug delivery. RSC Adv. 2020, 11, 442–451. [Google Scholar] [CrossRef]
- Usach, I.; Di Marco, S.; Díez, O.; Alós, M.; Peris, J.E. The Usefulness of In Vitro Percutaneous Absorption Experiments Applying the Infinite Dose Technique to Predict In Vivo Plasma Levels: Comparison of Model-Predicted and Observed Plasma Concentrations of Nortriptyline in Rats. Pharmaceutics 2022, 14, 1457. [Google Scholar] [CrossRef]
- Lau, W.M.; Ng, K.W. Finite and Infinite Dosing. In Percutaneous Penetration Enhancers Drug Penetration Into/Through the Skin: Methodology and General Considerations; Dragicevic, N., Maibach, H.I., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 35–44. [Google Scholar] [CrossRef]
- Dehdashtian, A.; Stringer, T.P.; Warren, A.J.; Mu, E.W.; Amirlak, B.; Shahabi, L. Anatomy and Physiology of the Skin. In Melanoma: A Modern Multidisciplinary Approach; Riker, A.I., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 15–26. [Google Scholar] [CrossRef]
- Cracowski, J.L.; Roustit, M. Human Skin Microcirculation. Compr. Physiol. 2020, 10, 1105–1154. [Google Scholar] [CrossRef]
- Calcutt, J.J.; Roberts, M.S.; Anissimov, Y.G. Modeling drug transport within the viable skin—A review. Expert Opin. Drug Metab. Toxicol. 2021, 17, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Law, R.M.; Ngo, M.A.; Maibach, H.I. Twenty Clinically Pertinent Factors/Observations for Percutaneous Absorption in Humans. Am. J. Clin. Dermatol. 2020, 21, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Leppert, W.; Malec-Milewska, M.; Zajaczkowska, R.; Wordliczek, J. Transdermal and Topical Drug Administration in the Treatment of Pain. Molecules 2018, 23, 681. [Google Scholar] [CrossRef] [PubMed]
- Gudin, J.; Nalamachu, S. Utility of lidocaine as a topical analgesic and improvements in patch delivery systems. Postgrad. Med. 2020, 132, 28–36. [Google Scholar] [CrossRef]
- Doss, N. Topical Corticosteroid Abuse: Africa Perspective. In A Treatise on Topical Corticosteroids in Dermatology: Use, Misuse and Abuse; Lahiri, K., Ed.; Springer: Singapore, 2018; pp. 229–235. [Google Scholar] [CrossRef]
- Wester, R.C.; Maibach, H.I. Cutaneous pharmacokinetics: 10 steps to percutaneous absorption. Drug Metab. Rev. 1983, 14, 169–205. [Google Scholar] [CrossRef]
- Tomalik-Scharte, D.; Lazar, A.; Meins, J.; Bastian, B.; Ihrig, M.; Wachall, B.; Jetter, A.; Tantcheva-Poór, I.; Mahrle, G.; Fuhr, U. Dermal absorption of permethrin following topical administration. Eur. J. Clin. Pharmacol. 2005, 61, 399–404. [Google Scholar] [CrossRef]
- Liu, X.; Yousef, S.; Anissimov, Y.G.; van der Hoek, J.; Tsakalozou, E.; Ni, Z.; Grice, J.E.; Roberts, M.S. Diffusion modelling of percutaneous absorption kinetics. Predicting urinary excretion from in vitro skin permeation tests (IVPT) for an infinite dose. Eur. J. Pharm. Biopharm. 2020, 149, 30–44. [Google Scholar] [CrossRef]
- Jeong, W.Y.; Kwon, M.; Choi, H.E.; Kim, K.S. Recent advances in transdermal drug delivery systems: A review. Biomater. Res. 2021, 25, 24. [Google Scholar] [CrossRef]
- Jin, X.; Imran, M.; Mohammed, Y. Topical Semisolid Products-Understanding the Impact of Metamorphosis on Skin Penetration and Physicochemical Properties. Pharmaceutics 2022, 14, 2487. [Google Scholar] [CrossRef]
- Zakaria, F.; Ashari, S.E.; Mat Azmi, I.D.; Abdul Rahman, M.B. Recent advances in encapsulation of drug delivery (active substance) in cubosomes for skin diseases. J. Drug Deliv. Sci. Technol. 2022, 68, 103097. [Google Scholar] [CrossRef]
- Noreen, S.; Ma, J.X.; Saeed, M.; Pervaiz, F.; Hanif, M.F.; Ahmed, B.; Farooq, M.I.; Akram, F.; Safdar, M.; Madni, A.; et al. Natural polysaccharide-based biodegradable polymeric platforms for transdermal drug delivery system: A critical analysis. Drug Deliv. Transl. Res. 2022, 12, 2649–2666. [Google Scholar] [CrossRef] [PubMed]
- Kellaway, I.W. The Influence of Formulation on Percutaneous Absorption. In Topics in Topicals: Current Trends in the Formulation of Topical Agents; Marks, R., Ed.; Springer: Dordrecht, The Netherlands, 1985; pp. 121–126. [Google Scholar] [CrossRef]
- Kuswahyuning, R.; Grice, J.E.; Moghimi, H.R.; Roberts, M.S. Formulation Effects in Percutaneous Absorption. In Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Drug Manipulation Strategies and Vehicle Effects; Dragicevic, N., Maibach, H.I., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 109–134. [Google Scholar] [CrossRef]
- Sobańska, A.W.; Brzezińska, E. IAM Chromatographic Models of Skin Permeation. Molecules 2022, 27, 1893. [Google Scholar] [CrossRef] [PubMed]
- da Costa Bernardo Port, B.; Schneider-Rauber, G.; Fretes Argenta, D.; Arhangelskis, M.; de Campos, C.E.M.; João Bortoluzzi, A.; Caon, T. Effect of Vehicle Composition on the Preparation of Different Types of Dapsone Crystals for Topical Drug Delivery. Mol. Pharm. 2022, 19, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Reddy, A.; Santra, M.; Khamanga, S. Amorphization of Drugs for Transdermal Delivery-a Recent Update. Pharmaceutics 2022, 14, 983. [Google Scholar] [CrossRef]
- Najib, O.N.; Martin, G.P.; Kirton, S.B.; Botha, M.J.; Sallam, A.S.; Murnane, D. The Influence of Oily Vehicle Composition and Vehicle-Membrane Interactions on the Diffusion of Model Permeants across Barrier Membranes. Membranes 2021, 11, 57. [Google Scholar] [CrossRef]
- Roberts, M.S.; Anderson, R.A. The percutaneous absorption of phenolic compounds: The effect of vehicles on the penetration of phenol. J. Pharm. Pharmacol. 1975, 27, 599–605. [Google Scholar] [CrossRef]
- Idson, B. Vehicle Effects in Percutaneous Absorption. Drug Metab. Rev. 1983, 14, 207–222. [Google Scholar] [CrossRef]
- Di Colo, G.; Carelli, V.; Giannaccini, B.; Serafini, M.F.; Bottari, F. Vehicle effects in percutaneous absorption: In vitro study of influence of solvent power and microscopic viscosity of vehicle on benzocaine release from suspension hydrogels. J. Pharm. Sci. 1980, 69, 387–391. [Google Scholar] [CrossRef]
- Tsakovska, I.; Pajeva, I.; Al Sharif, M.; Alov, P.; Fioravanzo, E.; Kovarich, S.; Worth, A.P.; Richarz, A.N.; Yang, C.; Mostrag-Szlichtyng, A.; et al. Quantitative structure-skin permeability relationships. Toxicology 2017, 387, 27–42. [Google Scholar] [CrossRef]
- Patel, H.; ten Berge, W.; Cronin, M.T. Quantitative structure-activity relationships (QSARs) for the prediction of skin permeation of exogenous chemicals. Chemosphere 2002, 48, 603–613. [Google Scholar] [CrossRef]
- Najib, O.N.; Kirton, S.B.; Martin, G.P.; Botha, M.J.; Sallam, A.S.; Murnane, D. Multivariate Analytical Approaches to Identify Key Molecular Properties of Vehicles, Permeants and Membranes That Affect Permeation through Membranes. Pharmaceutics 2020, 12, 958. [Google Scholar] [CrossRef] [PubMed]
- Queille-Roussel, C.; Nielsen, J.; Lacour, J.P. Vasoconstrictor potency of fixed-dose combination calcipotriol (50 μg/g) and betamethasone dipropionate (0.5 mg/g) cutaneous foam versus other topical corticosteroids used to treat psoriasis vulgaris. J. Dermatol. Treat. 2019, 30, 529–533. [Google Scholar] [CrossRef]
- Queille-Roussel, C.; Bang, B.; Clonier, F.; Lacour, J.P. Enhanced vasoconstrictor potency of the fixed combination calcipotriol plus betamethasone dipropionate in an innovative aerosol foam formulation vs. other corticosteroid psoriasis treatments. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1951–1956. [Google Scholar] [CrossRef]
- Hattori, T.; Tagawa, H.; Inai, M.; Kan, T.; Kimura, S.I.; Itai, S.; Mitragotri, S.; Iwao, Y. Transdermal delivery of nobiletin using ionic liquids. Sci. Rep. 2019, 9, 20191. [Google Scholar] [CrossRef]
- Kandregula, B.; Narisepalli, S.; Chitkara, D.; Mittal, A. Exploration of Lipid-Based Nanocarriers as Drug Delivery Systems in Diabetic Foot Ulcer. Mol. Pharm. 2022, 19, 1977–1998. [Google Scholar] [CrossRef]
- Wurster, D.E.; Kramer, S.F. Investigation of Some Factors Influencing Percutaneous Absorption. J. Pharm. Sci. 1961, 50, 288–293. [Google Scholar] [CrossRef]
- Angamuthu, M.; Shankar, V.K.; Murthy, S.N. Water Activity and Its Significance in Topical Dosage Forms. J. Pharm. Sci. 2018, 107, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Yousef, S.; Mohammed, Y.; Namjoshi, S.; Grice, J.; Sakran, W.; Roberts, M. Mechanistic Evaluation of Hydration Effects on the Human Epidermal Permeation of Salicylate Esters. AAPS J. 2017, 19, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Telaprolu, K.C.; Grice, J.E.; Mohammed, Y.H.; Roberts, M.S. Human Skin Drug Metabolism: Relationships between Methyl Salicylate Metabolism and Esterase Activities in IVPT Skin Membranes. Metabolites 2023, 13, 934. [Google Scholar] [CrossRef]
- Ossowicz, P.; Klebeko, J.; Janus, E.; Nowak, A.; Duchnik, W.; Kucharski, Ł.; Klimowicz, A. The effect of alcohols as vehicles on the percutaneous absorption and skin retention of ibuprofen modified with l-valine alkyl esters. RSC Adv. 2020, 10, 41727–41740. [Google Scholar] [CrossRef]
- Akram, M.W.; Jamshaid, H.; Rehman, F.U.; Zaeem, M.; Khan, J.z.; Zeb, A. Transfersomes: A Revolutionary Nanosystem for Efficient Transdermal Drug Delivery. AAPS PharmSciTech 2021, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Barbalho, G.N.; Brugger, S.; Raab, C.; Lechner, J.-S.; Gratieri, T.; Keck, C.M.; Rupenthal, I.D.; Agarwal, P. Development of transferosomes for topical ocular drug delivery of curcumin. Eur. J. Pharm. Biopharm. 2024, 205, 114535. [Google Scholar] [CrossRef] [PubMed]
- Chamel, S.; Mishra, A.; Gull, A. Transferosomes as innovative drug delivery systems for enhanced antifungal therapy: A comprehensive review. J. Drug Deliv. Sci. Technol. 2024, 95, 105545. [Google Scholar] [CrossRef]
- Tapfumaneyi, P.; Imran, M.; Mohammed, Y.; Roberts, M.S. Recent advances and future prospective of topical and transdermal delivery systems. Front. Drug Deliv. 2022, 2, 957732. [Google Scholar] [CrossRef]
- Niu, X.Q.; Zhang, D.P.; Bian, Q.; Feng, X.F.; Li, H.; Rao, Y.F.; Shen, Y.M.; Geng, F.N.; Yuan, A.R.; Ying, X.Y.; et al. Mechanism investigation of ethosomes transdermal permeation. Int. J. Pharm. X 2019, 1, 100027. [Google Scholar] [CrossRef]
- Kumar Sahu, A.K.; Kashyap, P.; Mishra, K.; Mishra, S.P.; Dash, D.K.; Kaur, C.D.; Verma, S. Transethosomes and Nanoethosomes: Recent Approach on Transdermal Drug Delivery System. In Nanomedicines; Farrukh, M.A., Ed.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar] [CrossRef]
- Sudhakar, K.; Fuloria, S.; Subramaniyan, V.; Sathasivam, K.; Azad, A.K.; Swain, S.; Sekar, M.; Karupiah, S.; Porwal, O.; Sahoo, A.; et al. Ultraflexible Liposome Nanocargo as a Dermal and Transdermal Drug Delivery System. Nanomaterials 2021, 2021, 2557. [Google Scholar] [CrossRef]
- Zoabi, A.; Touitou, E.; Margulis, K. Recent Advances in Nanomaterials for Dermal and Transdermal Applications. Colloids Interfaces 2021, 5, 18. [Google Scholar] [CrossRef]
- Sala, M.; Diab, R.; Elaissari, A.; Fessi, H. Lipid nanocarriers as skin drug delivery systems: Properties, mechanisms of skin interactions and medical applications. Int. J. Pharm. 2018, 535, 1–17. [Google Scholar] [CrossRef]
- Waghule, T.; Singhvi, G.; Dubey, S.K.; Pandey, M.M.; Gupta, G.; Singh, M.; Dua, K. Microneedles: A smart approach and increasing potential for transdermal drug delivery system. Biomed. Pharmacother. 2019, 109, 1249–1258. [Google Scholar] [CrossRef]
- Yu, Y.Q.; Yang, X.; Wu, X.F.; Fan, Y.B. Enhancing Permeation of Drug Molecules Across the Skin via Delivery in Nanocarriers: Novel Strategies for Effective Transdermal Applications. Front. Bioeng. Biotechnol. 2021, 9, 646554. [Google Scholar] [CrossRef]
- Bos, J.D.; Meinardi, M.M. The 500 Dalton rule for the skin penetration of chemical compounds and drugs. Exp. Dermatol. 2000, 9, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Ghosh, P.; Li, S.K.; Newman, B.; Kasting, G.B.; Raney, S.G. Heat effects on drug delivery across human skin. Expert. Opin. Drug Deliv. 2016, 13, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Hull, W. Heat-enhanced transdermal drug delivery: A survey paper. J. Appl. Res. 2002, 2, XXIII–XXIV. [Google Scholar]
- La Count, T.D.; Zhang, Q.; Murawsky, M.; Hao, J.; Ghosh, P.; Dave, K.; Raney, S.G.; Talattof, A.; Kasting, G.B.; Li, S.K. Evaluation of Heat Effects on Transdermal Nicotine Delivery In Vitro and In Silico Using Heat-Enhanced Transport Model Analysis. AAPS J. 2020, 22, 82. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Tonnis, K.; Xu, L.; Jaworska, J.; Kasting, G.B. Modeling the Percutaneous Absorption of Solvent-deposited Solids Over a Wide Dose Range. J. Pharm. Sci. 2022, 111, 769–779. [Google Scholar] [CrossRef]
- Kathe, K.; Kathpalia, H. Film forming systems for topical and transdermal drug delivery. Asian J. Pharm. Sci. 2017, 12, 487–497. [Google Scholar] [CrossRef]
- Pünnel, L.C.; Lunter, D.J. Film-Forming Systems for Dermal Drug Delivery. Pharmaceutics 2021, 13, 932. [Google Scholar] [CrossRef]
- Šnejdrová, E.; Martiška, J.; Loskot, J.; Paraskevopoulos, G.; Kováčik, A.; Regdon, G., Jr.; Budai-Szűcs, M.; Palát, K.; Konečná, K. PLGA based film forming systems for superficial fungal infections treatment. Eur. J. Pharm. Sci. 2021, 163, 105855. [Google Scholar] [CrossRef]
- Yamada, M.; Mohammed, Y.; Prow, T.W. Advances and controversies in studying sunscreen delivery and toxicity. Adv. Drug Deliv. Rev. 2020, 153, 72–86. [Google Scholar] [CrossRef]
- Surber, C.; Knie, U. Metamorphosis of Vehicles: Mechanisms and Opportunities. Curr. Probl. Dermatol. 2018, 54, 152–165. [Google Scholar] [CrossRef]
- Gennari, C.G.M.; Selmin, F.; Minghetti, P.; Cilurzo, F. Medicated Foams and Film Forming Dosage Forms as Tools to Improve the Thermodynamic Activity of Drugs to be Administered Through the Skin. Curr. Drug Deliv. 2019, 16, 461–471. [Google Scholar] [CrossRef]
- Stein Gold, L.; Paul, C.; Romiti, R. Efficacy and safety of fixed-dose combination calcipotriol/betamethasone dipropionate foam for the treatment of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Selmin, F.; Franzè, S.; Casiraghi, A.; Cilurzo, F. Spotlight on Calcipotriol/Betamethasone Fixed-Dose Combination in Topical Formulations: Is There Still Room for Innovation? Pharmaceutics 2022, 14, 2085. [Google Scholar] [CrossRef] [PubMed]
- Danby, S.G.; Draelos, Z.D.; Gold, L.F.S.; Cha, A.; Vlahos, B.; Aikman, L.; Sanders, P.; Wu-Linhares, D.; Cork, M.J. Vehicles for atopic dermatitis therapies: More than just a placebo. J. Dermatolog Treat. 2022, 33, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Thakur, A.; Mandal, U.K.; Thakur, A.; Bhatia, A. Foam-Based Drug Delivery: A Newer Approach for Pharmaceutical Dosage Form. AAPS PharmSciTech 2022, 23, 244. [Google Scholar] [CrossRef] [PubMed]
- Kircik, L.; Del Rosso, J.Q.; Weiss, J.S.; Stakias, V.; London, A.; Keynan, R.; Hazot, Y.; Elliott, R.; Stuart, I. Formulation and Profile of FMX101 4% Minocycline Topical Foam for the Treatment of Acne Vulgaris. J. Clin. Aesthet. Dermatol. 2020, 13, 14–21. [Google Scholar]
- Van Bocxlaer, K.; McArthur, K.N.; Harris, A.; Alavijeh, M.; Braillard, S.; Mowbray, C.E.; Croft, S.L. Film-Forming Systems for the Delivery of DNDI-0690 to Treat Cutaneous Leishmaniasis. Pharmaceutics 2021, 13, 516. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tapfumaneyi, P.; Phan, K.; Huang, Y.; Sodsri, K.; Namjoshi, S.; Maibach, H.; Mohammed, Y. Solute–Vehicle–Skin Interactions and Their Contribution to Pharmacokinetics of Skin Delivery. Pharmaceutics 2025, 17, 764. https://doi.org/10.3390/pharmaceutics17060764
Tapfumaneyi P, Phan K, Huang Y, Sodsri K, Namjoshi S, Maibach H, Mohammed Y. Solute–Vehicle–Skin Interactions and Their Contribution to Pharmacokinetics of Skin Delivery. Pharmaceutics. 2025; 17(6):764. https://doi.org/10.3390/pharmaceutics17060764
Chicago/Turabian StyleTapfumaneyi, Pronalis, Khanh Phan, Yicheng Huang, Kewaree Sodsri, Sarika Namjoshi, Howard Maibach, and Yousuf Mohammed. 2025. "Solute–Vehicle–Skin Interactions and Their Contribution to Pharmacokinetics of Skin Delivery" Pharmaceutics 17, no. 6: 764. https://doi.org/10.3390/pharmaceutics17060764
APA StyleTapfumaneyi, P., Phan, K., Huang, Y., Sodsri, K., Namjoshi, S., Maibach, H., & Mohammed, Y. (2025). Solute–Vehicle–Skin Interactions and Their Contribution to Pharmacokinetics of Skin Delivery. Pharmaceutics, 17(6), 764. https://doi.org/10.3390/pharmaceutics17060764