
Influence of Ionization and the Addition of Cyclodextrins and Hydrophilic Excipients on the Solubility of Benzthiazide, Isoxicam, and Piroxicam





Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Instruments
2.2.1. Shake-Flask Method
2.2.2. CheqSol Method
2.3. Procedures
2.3.1. pKa and logPo/w Determination
2.3.2. Shake-Flask Solubility Determination
2.3.3. CheqSol Solubility Determination
3. Results and Discussion
3.1. Shake-Flask Determinations
3.2. CheqSol Determinations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| API | Active pharmaceutical ingredient |
| EXC | Excipient |
| CAV | Cavasol |
| CAP | Captisol |
| KLU | Klucel |
| KOL | Kollidon |
| S630 | Plasdone S630 |
References
- Avdeef, A. Absorption and Drug Development: Solubility, Permeability, and Charge State, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 9781118057452. [Google Scholar]
- Di, L. Drug-like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization, 2nd ed.; Kerns, E.H., Ed.; Academic Press: Amsterdam, The Netherlands, 2016; ISBN 0128013222. [Google Scholar]
- Völgyi, G.; Box, K.J.; Comer, J.E.A.; Takács-Novák, K. Study of PH-Dependent Solubility of Organic Bases. Revisit of Henderson-Hasselbalch Relationship. Anal. Chim. Acta 2010, 673, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Baka, E.; Comer, J.E.A.; Takács-Novák, K. Study of Equilibrium Solubility Measurement by Saturation Shake-Flask Method Using Hydrochlorothiazide as Model Compound. J. Pharm. Biomed. Anal. 2008, 46, 335–341. [Google Scholar] [CrossRef]
- Hsieh, Y.L.; Ilevbare, G.A.; Eerdenbrugh, B.; Box, K.J.; Sanchez-Felix, M.V.; Taylor, L.S. pH-induced precipitation behavior of weakly basic compounds: Determination of extent and duration of supersaturation using potentiometric titration and correlation to solid sstate properties. Pharm. Res. 2012, 29, 2738–2753. [Google Scholar] [CrossRef] [PubMed]
- Shoghi, E.; Fuguet, E.; Bosch, E.; Ràfols, C. Solubility-PH Profiles of Some Acidic, Basic and Amphoteric Drugs. Eur. J. Pharm. Sci. 2013, 48, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of In Vitro Drug Product Dissolution and In Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER). Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System; Guidance for Industry; Center for Drug Evaluation and Research: Silver Spring, MD, USA, 2017. [Google Scholar]
- Kostewicz, E.S.; Wunderlich, M.; Brauns, U.; Becker, R.; Bock, T.; Dressman, J.B. Predicting the Precipitation of Poorly Soluble Weak Bases upon Entry in the Small Intestine. J. Pharm. Pharmacol. 2004, 56, 43–51. [Google Scholar] [CrossRef]
- Fornells, E.; Fuguet, E.; Mañé, M.; Ruiz, R.; Box, K.; Bosch, E.; Ràfols, C. Effect of Vinylpyrrolidone Polymers on the Solubility and Supersaturation of Drugs; A Study Using the Cheqsol Method. Eur. J. Pharm. Sci. 2018, 117, 227–235. [Google Scholar] [CrossRef]
- Van Duong, T.; Van Den Mooter, G. The Role of the Carrier in the Formulation of Pharmaceutical Solid Dispersions. Part II: Amorphous Carriers. Expert Opin. Drug Deliv. 2016, 13, 1681–1694. [Google Scholar] [CrossRef]
- Van Duong, T.; Ni, Z.; Taylor, L.S. Phase Behavior and Crystallization Kinetics of a Poorly Water-Soluble Weakly Basic Drug as a Function of Supersaturation and Media Composition. Mol. Pharm. 2022, 19, 1146–1159. [Google Scholar] [CrossRef]
- Narang, A.S.; Boddu, S.H. Excipient Applications in Formulation Design and Drug Delivery; Springer: Cham, Switzerland, 2015; pp. 1–681. ISBN 978-3-319-20205-1. [Google Scholar] [CrossRef]
- Loftsson, T.; Duchêne, D. Historical Perspectives Cyclodextrins and Their Pharmaceutical Applications. Int. J. Pharm. 2007, 329, 1–11. [Google Scholar] [CrossRef]
- Warren, D.B.; Benameur, H.; Porter, C.J.H.; Pouton, C.W. Using Polymeric Precipitation Inhibitors to Improve the Absorption of Poorly Water-Soluble Drugs: A Mechanistic Basis for Utility. J. Drug Target. 2010, 18, 704–731. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Jarho, P.; Másson, M.; Järvinen, T. Cyclodextrins in Drug Delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in Drug Delivery: An Updated Review. AAPS PharmSciTech 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, Physicochemical Properties and Pharmaceutical Applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Loftsson, T.; Hreinsdóttir, D.; Másson, M. Evaluation of Cyclodextrin Solubilization of Drugs. Int. J. Pharm. 2005, 302, 18–28. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in Pharmaceutical Formulations II: Solubilization, Binding Constant, and Complexation Efficiency. Drug Discov. Today 2016, 21, 363–368. [Google Scholar] [CrossRef]
- Jagtap, P.S.; Tagad, R.R.; Shendge, R.S. A Brief Review on Kollidon. J. Drug Deliv. Ther. 2019, 9, 493–500. [Google Scholar] [CrossRef]
- Nikghalb, L.A.; Singh, G.; Singh, G.; Kahkeshan, K.F. Solid Dispersion: Methods and Polymers to Increase the Solubility of Poorly Soluble Drugs. J. Appl. Pharm. Sci. 2012, 2, 170–175. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving Drug Solubility for Oral Delivery Using Solid Dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Ghasemi-Asl, S.; Shayanfa, A. The effect of pH and beta-cyclodextrin on solubility and solution stability of piroxicam cocrystals. J. Mol. Liq. 2025, 422, 126936. [Google Scholar] [CrossRef]
- Lucero-Borja, D.; Subirats, X.; Barbas, R.; Prohens, R.; Avdeef, A.; Ràfols, C. Potentiometric CheqSol and Standardized Shake-Flask Solubility Methods Are Complimentary Tools in Physicochemical Profiling. Eur. J. Pharm. Sci. 2020, 148, 105305. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A.; Fuguet, E.; Llinàs, A.; Ràfols, C.; Bosch, E.; Völgyi, G.; Verbic, T.; Boldyreva, E.; Takács-Novák, K. Equilibrium Solubility Measurement of Ionizable Drugs—Consensus Recommendations for Improving Data Quality. ADMET DMPK 2016, 4, 117–178. [Google Scholar] [CrossRef]
- Stuart, M.; Box, K. Chasing Equilibrium: Measuring the Intrinsic Solubility of Weak Acids and Bases. Anal. Chem. 2005, 77, 983–990. [Google Scholar] [CrossRef] [PubMed]
- ACD Percepta Platform, version 2012; Advanced Chemistry Development, Inc. (ACD/Labs): Toronto, ON, Canada, 2012. Available online: www.acdlabs.com (accessed on 18 March 2025).
- Avdeef, A.; Bucher, J.J. Accurate Measurements of the Concentration of Hydrogen Ions with a Glass Electrode: Calibrations Using the Prideaux and Other Universal Buffer Solutions and a Computer-Controlled Automatic Titrator. Anal. Chem. 1978, 50, 2137–2142. [Google Scholar] [CrossRef]
- Allen, R.I.; Box, K.J.; Comer, J.E.A.; Peake, C.; Tam, K.Y. Multiwavelength Spectrophotometric Determination of Acid Dissociation Constants of Ionizable Drugs. J. Pharm. Biomed. Anal. 1998, 17, 699–712. [Google Scholar] [CrossRef]
- Avdeef, A. PH-Metric LogP. II: Refinement of Partition Coefficients and Lonization Constants of Multiprotic Substances. J. Pharm. Sci. 1993, 82, 183–190. [Google Scholar] [CrossRef]
- Avdeef, A.; Comer, J.E.A.; Thomson, S.J. PH-Metric Log P. 3. Glass Electrode Calibration in Methanol-Water, Applied to pKa Determination of Water-Insoluble Substances. Anal. Chem. 1993, 65, 42–49. [Google Scholar] [CrossRef]
- Biorelevant. Available online: www.biorelevant.com (accessed on 18 March 2025).
- Avdeef, A. Multi-Lab Intrinsic Solubility Measurement Reproducibility in CheqSol and Shake-Flask Methods. ADMET DMPK 2019, 7, 210–219. [Google Scholar] [CrossRef]
- Goswami, S.; Majumdar, A.; Sarkar, M. Painkiller Isoxicam and Its Copper Complex Can Form Inclusion Complexes with Different Cyclodextrins: A Fluorescence, Fourier Transform Infrared Spectroscopy, and Nuclear Magnetic Resonance Study. J. Phys. Chem. B 2017, 121, 8454–8466. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | pKa | log Po/w | Molar Volume 2 (cm3) |
|---|---|---|---|---|
| Benzthiazide | 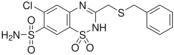 | 6.64 1; 9.22 1 | 1.89 (0.02) | 259.0 |
| Isoxicam | 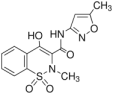 | 3.84 1 | 2.99 (0.02) | 211.0 |
| Piroxicam | 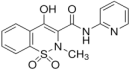 | 1.89 (0.01); 5.34 (0.01) | 1.71 (0.02) | 211.9 |
| Excipient | SEXC/SAPI | |||
|---|---|---|---|---|
| Benzthiazide | Isoxicam | Piroxicam | ||
| pH 2 1 | Captisol | 1.14 | 1.48 | 1.44 |
| Cavasol | 0.97 | 1.45 | 1.12 | |
| Klucel | 1.20 | 1.50 | 1.94 | |
| Kollidon | 1.29 | 1.47 | 1.54 | |
| S630 | 1.61 | 1.81 | 2.02 | |
| pH 3.5 2 | Captisol | --- | --- | 1.27 |
| Cavasol | --- | --- | 0.96 | |
| Klucel | --- | --- | 1.69 | |
| Kollidon | --- | --- | 1.55 | |
| S630 | --- | --- | 2.19 | |
| pH 5.8 | Captisol | 1.09 | 1.44 | 0.96 |
| Cavasol | 0.83 | 1.56 | 1.25 | |
| Klucel | 1.12 | 1.93 | 1.73 | |
| Kollidon | 1.21 | 1.88 | 1.70 | |
| S630 | 1.49 | 1.96 | 1.99 | |
| pH 6.5 | Captisol | 1.29 | 1.52 | 1.03 |
| Cavasol | 0.98 | 1.61 | 1.56 | |
| Klucel | 1.15 | 1.84 | 2.01 | |
| Kollidon | 1.24 | 1.93 | 2.24 | |
| S630 | 1.28 | 1.73 | 2.92 | |
| Compound–Excipient | Cmax (µM) | ts (min) | Rs |
|---|---|---|---|
| Benzthiazide | 44 (11) | 6 (1) | 4 (1) |
| Benzthiazide–Captisol | 50 (5) | 6 (1) | 4.2 (0.4) |
| Benzthiazide–Cavasol | 92 (10) | 7 (1) | 7.7 (0.8) |
| Benzthiazide–Klucel | 516 (54) | 14 (2) | 43 (5) |
| Benzthiazide–Kollidon | 1404 (178) | 21 (1) | 104 (26) |
| Benzthiazide–Plasdone S630 | 535 (87) | 25 (3) | 45 (8) |
| Isoxicam | 92 (9) | 11 (3) | 48 (4) |
| Isoxicam–Captisol | 137 (6) | 8 (1) | 71 (3) |
| Isoxicam–Cavasol | 122 (12) | 7 (1) | 64 (6) |
| Isoxicam–Klucel | 195 (13) | 16 (6) | 102 (7) |
| Isoxicam–Kollidon | 317 (73) | 18 (4) | 166 (38) |
| Isoxicam–Plasdone S630 | 200 (18) | 19 (3) | 105 (9) |
| Piroxicam | 706 (135) | 7 (1) | 34 (6) |
| Piroxicam–Captisol | 831 (56) | 8 (1) | 40 (3) |
| Piroxicam–Cavasol | 849 (130) | 7 (1) | 41 (6) |
| Piroxicam–Klucel | 2431 (454) | 13 (2) | 117 (22) |
| Piroxicam–Kollidon | 1980 (450) | 7 (1) | 95 (22) |
| Piroxicam–Plasdone S630 | 3561 (576) | 10 (1) | 171 (28) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucero-Borja, D.; Ruiz, R.; Fuguet, E.; Ràfols, C. Influence of Ionization and the Addition of Cyclodextrins and Hydrophilic Excipients on the Solubility of Benzthiazide, Isoxicam, and Piroxicam. Pharmaceutics 2025, 17, 571. https://doi.org/10.3390/pharmaceutics17050571
Lucero-Borja D, Ruiz R, Fuguet E, Ràfols C. Influence of Ionization and the Addition of Cyclodextrins and Hydrophilic Excipients on the Solubility of Benzthiazide, Isoxicam, and Piroxicam. Pharmaceutics. 2025; 17(5):571. https://doi.org/10.3390/pharmaceutics17050571
Chicago/Turabian StyleLucero-Borja, Diego, Rebeca Ruiz, Elisabet Fuguet, and Clara Ràfols. 2025. "Influence of Ionization and the Addition of Cyclodextrins and Hydrophilic Excipients on the Solubility of Benzthiazide, Isoxicam, and Piroxicam" Pharmaceutics 17, no. 5: 571. https://doi.org/10.3390/pharmaceutics17050571
APA StyleLucero-Borja, D., Ruiz, R., Fuguet, E., & Ràfols, C. (2025). Influence of Ionization and the Addition of Cyclodextrins and Hydrophilic Excipients on the Solubility of Benzthiazide, Isoxicam, and Piroxicam. Pharmaceutics, 17(5), 571. https://doi.org/10.3390/pharmaceutics17050571