The Molecular Chaperone TCP1 Affects Carcinogenicity and Is a Potential Therapeutic Target for Acute Myeloid Leukemia

 , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Cell Lines
2.3. Compounds and Reagents
2.4. Different Generations of Xenograft Tumor Models
2.5. Proteomic Analysis
2.6. Quantitative Real-Time PCR Analysis
- TCP1-Forward: AGCCACGCTATCCAGTCAAC;
- TCP1-Reverse: GGCTGAAGTCAAGGCAAGCA;
- hsa-miR-340-5p-Forward: GCCGCGCTTATAAAGCAATGAGACTGA.
2.7. Western Blot Analysis
2.8. Lentivirus Transfection
2.9. Protein Profile Analysis
2.10. Screen and Selection of the miRNA
2.11. Luciferase Reporter Gene Assay
2.12. SYBYL Software Analysis
2.13. MTT Assay
2.14. Surface Plasmon Resonance Analysis
2.15. Cell Colony Assay
2.16. Apoptosis Analysis
2.17. Cell Cycle Analysis
2.18. Co-Immunoprecipitation Analysis
2.19. The Effect of FTY720 on Xenograft AML Tumors
2.20. Statistical Analysis
3. Results
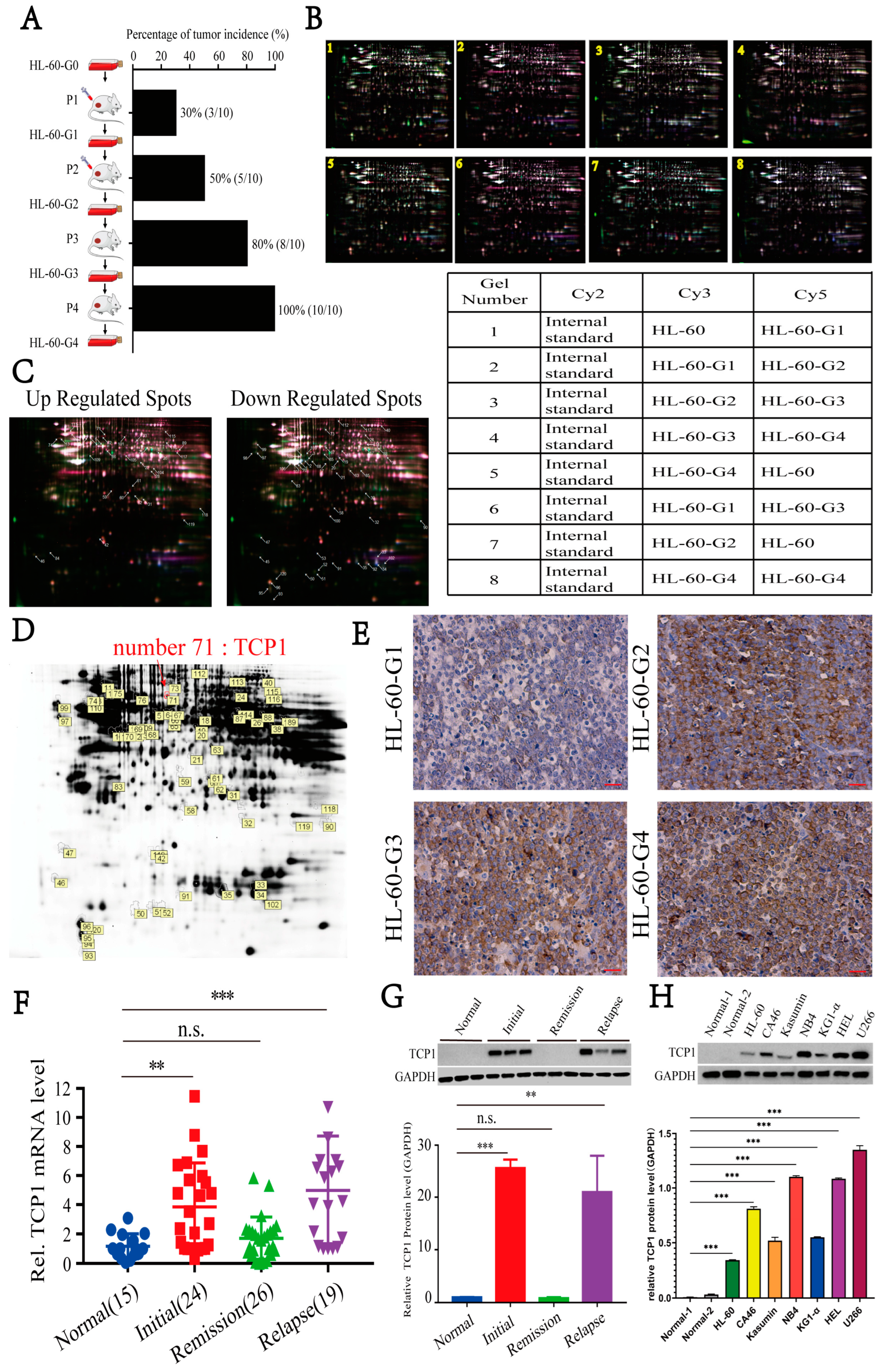
3.1. Elevated TCP1 Expression in Successive Xenograft AML Tumor Generations Correlates with HL-60 Cell Tumorigenicity
3.2. TCP1 May Regulate Multiple Signaling Pathways Related to AML Cell Survival
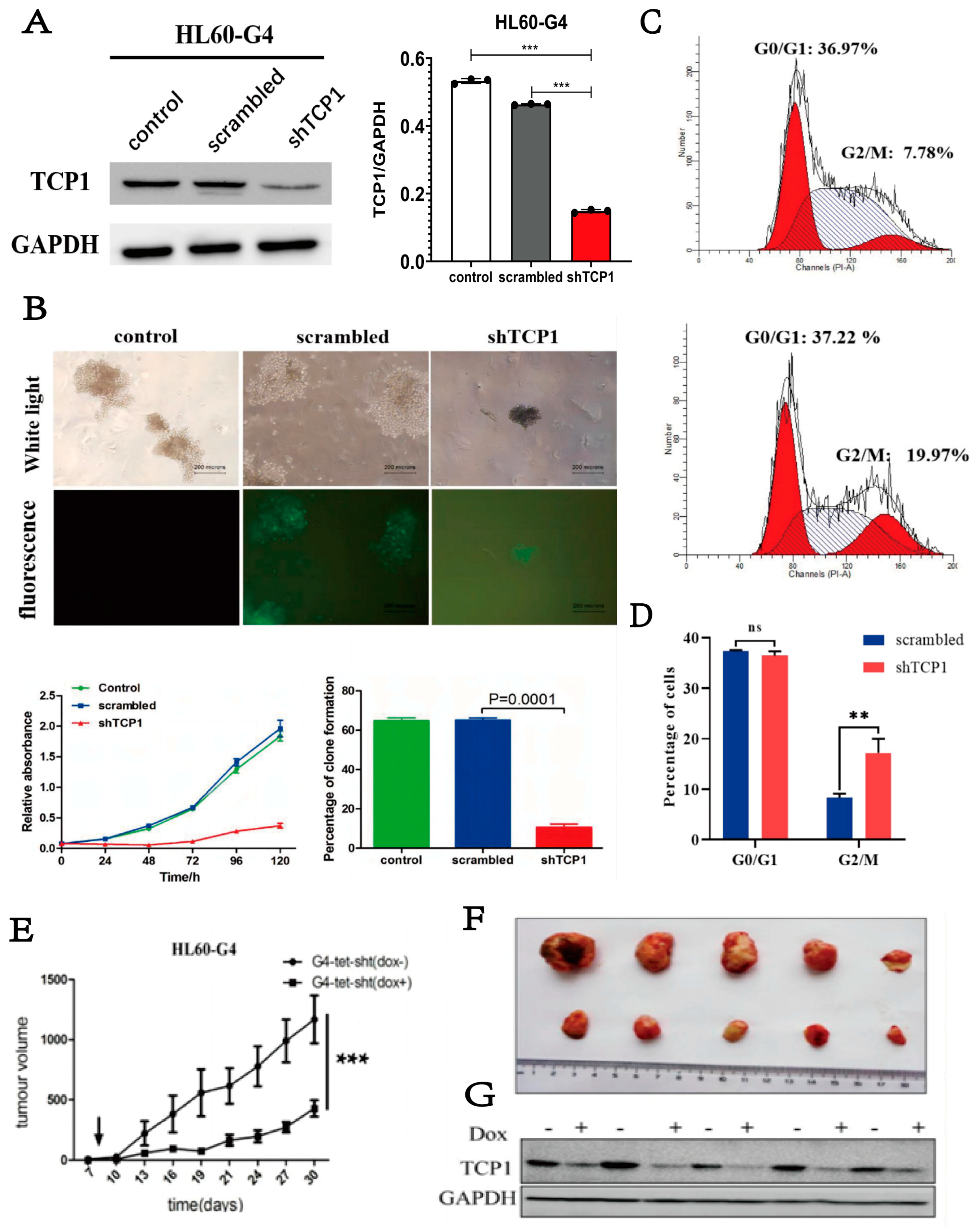
3.3. Knocking Down TCP1 Leads to Effective Inhibition of AML Cell Proliferation In Vitro and In Vivo
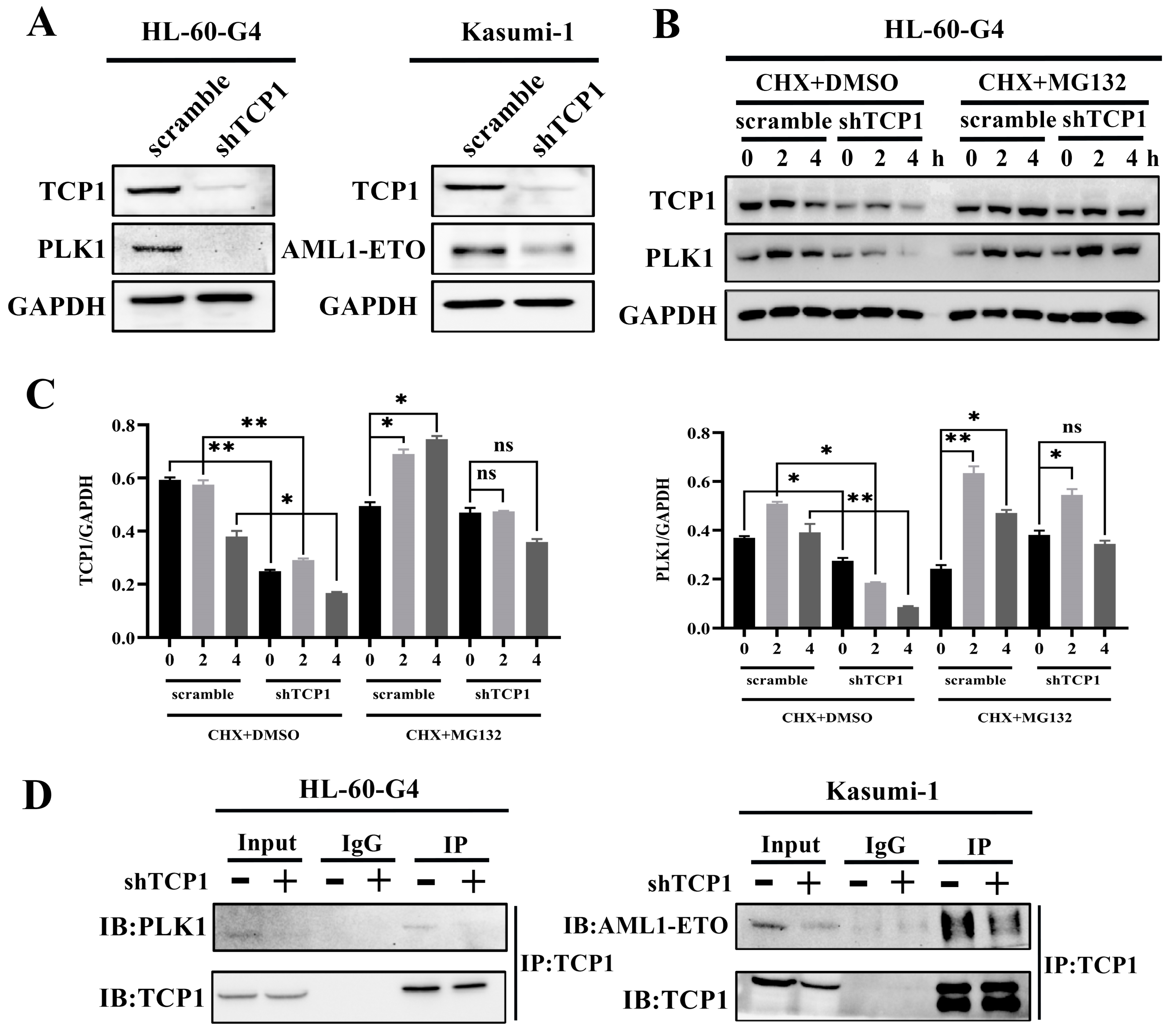
3.4. TCP1 Was Able to Maintain the Stability of the PLK1 and AML1-ETO Proteins Through Its Chaperone Function
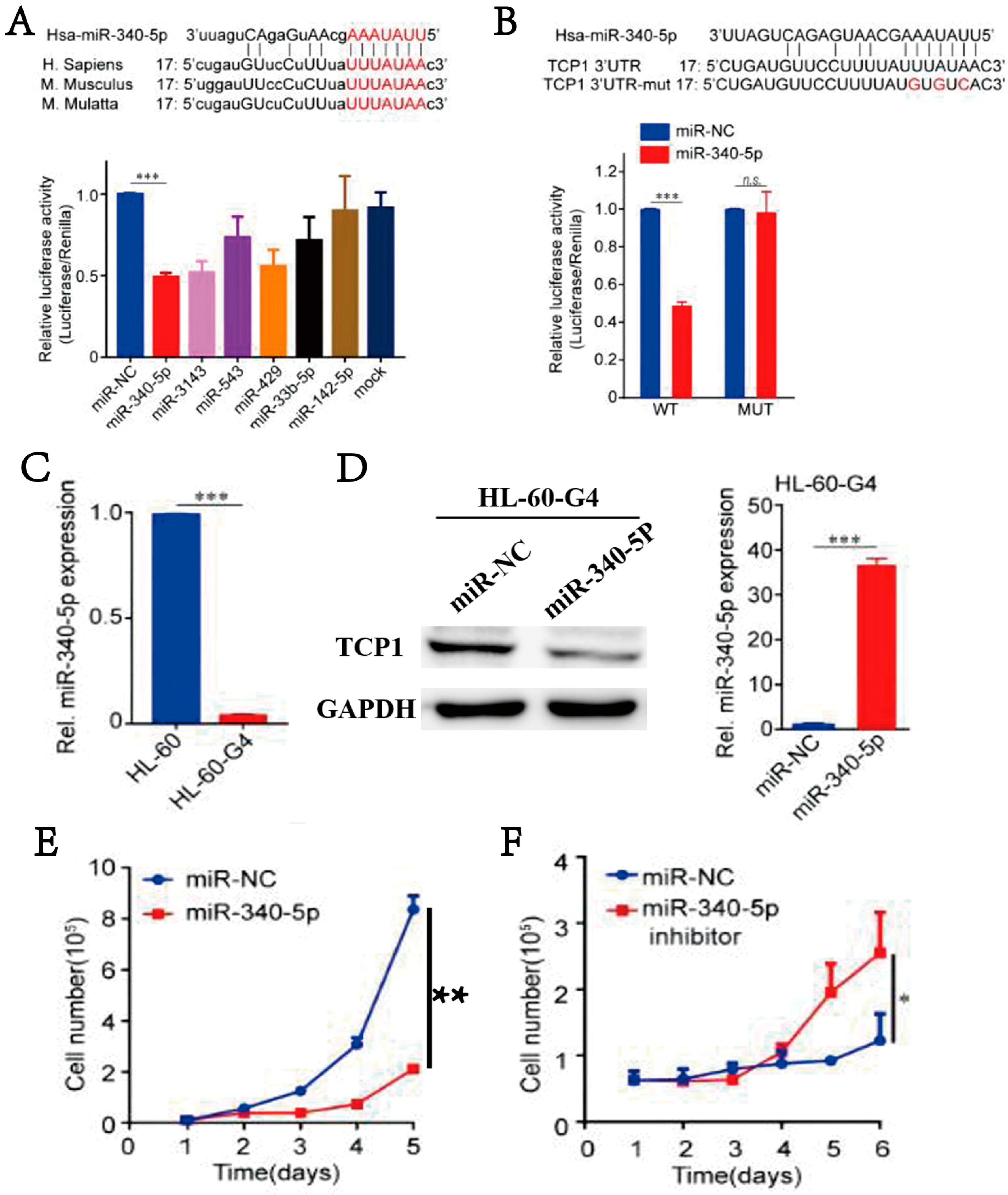
3.5. MiR-340-5p Can Inhibit the Expression of TCP1, Thereby Blocking AML Cell Proliferation
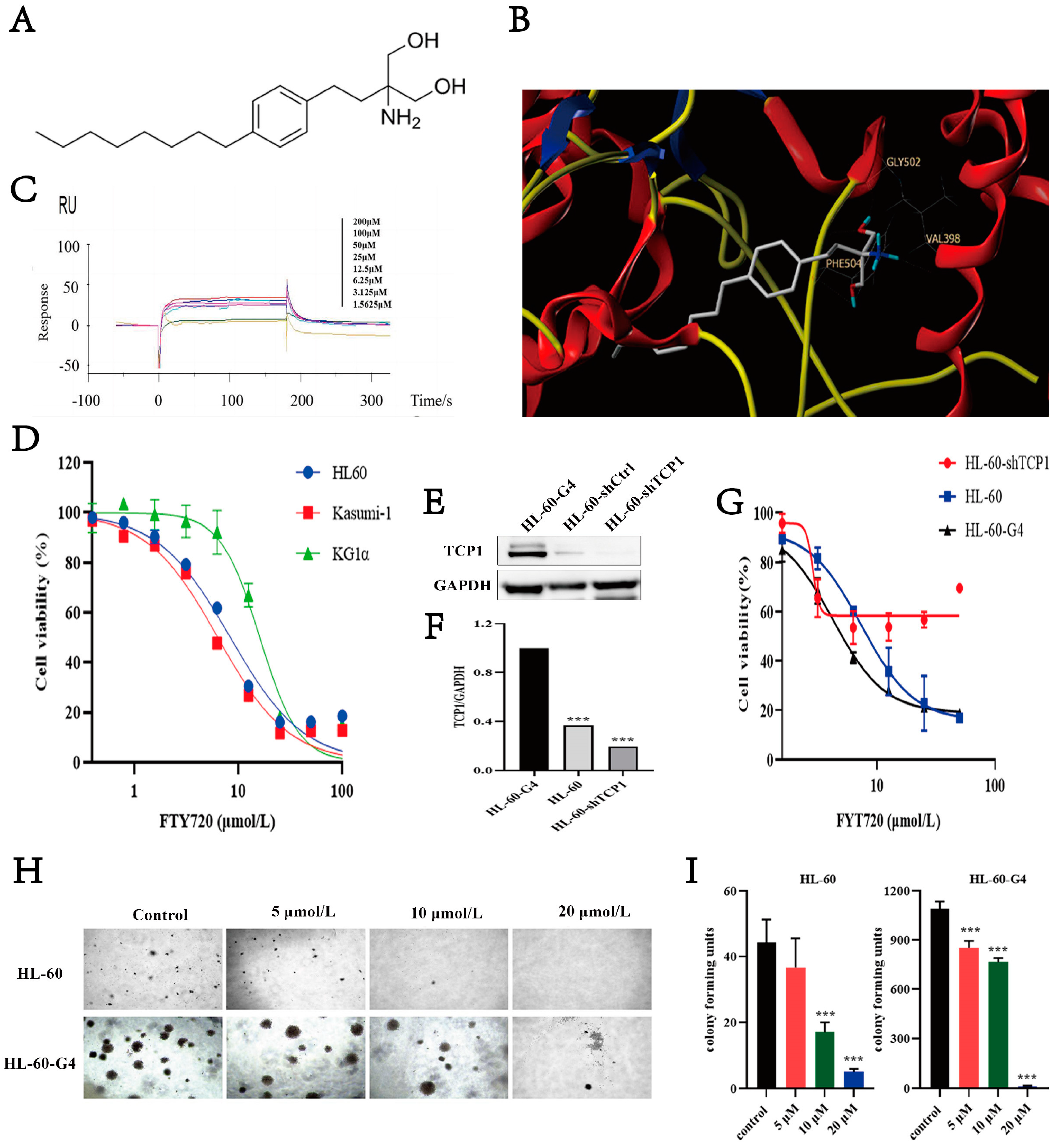
3.6. FTY720 Has an Affinity for TCP1 and Can Inhibit AML Cell Proliferation
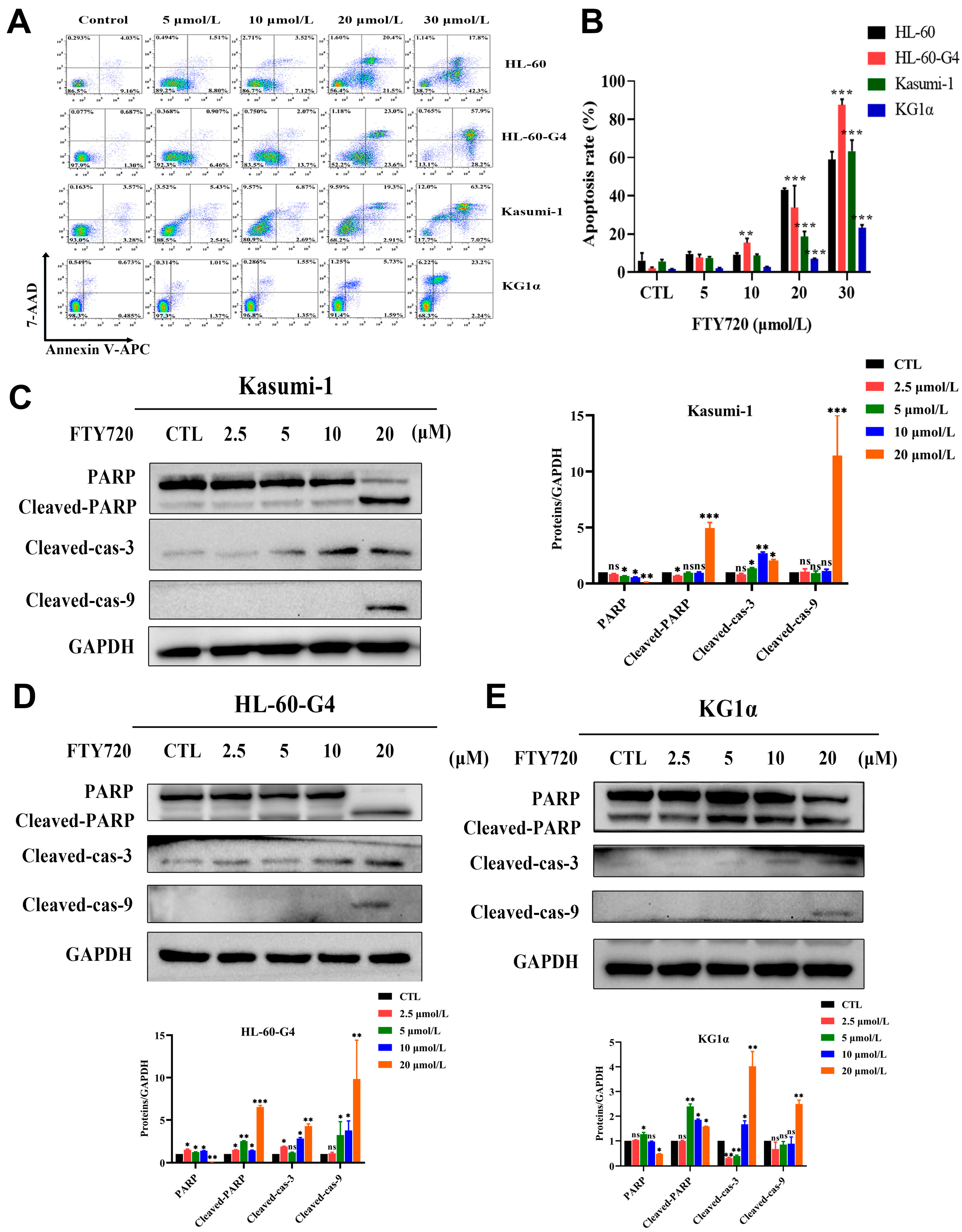
3.7. FTY720 Could Induce Apoptosis and Cell Cycle Arrest in AML Cells
3.8. FTY720 Decreased Protein Levels by Inhibiting TCP1 Chaperone Function
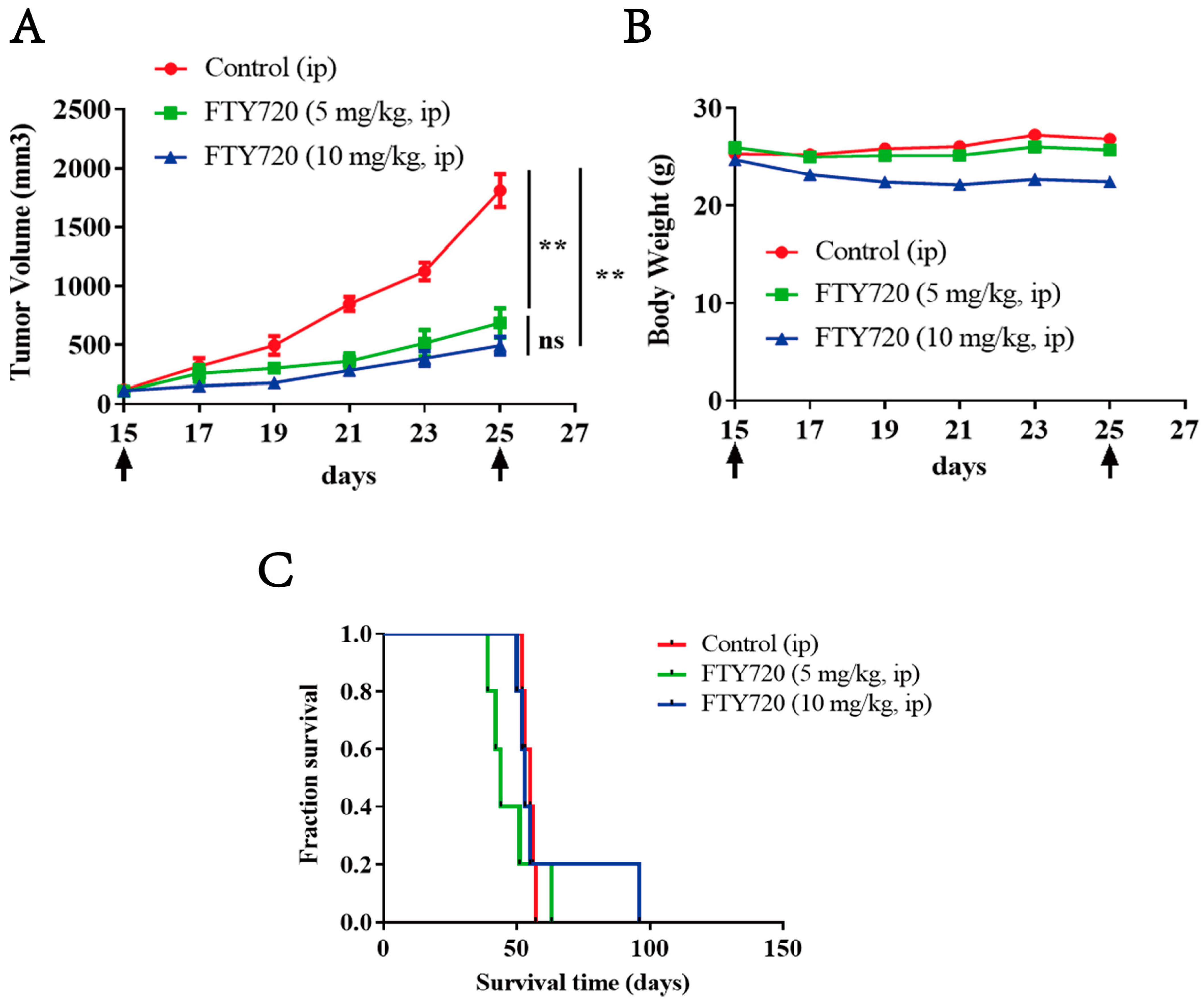
3.9. FTY720 Inhibited the Growth of Xenograft AML Tumors in Vivo and Extended the Survival Time of Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Vadakekolathu, J.; Rutella, S. Escape from T-cell–targeting immunotherapies in acute myeloid leukemia. Blood 2024, 143, 2689–2700. [Google Scholar] [CrossRef] [PubMed]
- Shimony, S.; Stahl, M.; Stone, R.M. Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, R.S.; Pratz, K.W.; Lai, C. Recent advances in targeted therapies in acute myeloid leukemia. J. Hematol. Oncol. 2023, 16, 29. [Google Scholar] [CrossRef]
- Chen, X.; Chen, X.; Huang, Y.; Lin, J.; Wu, Y.; Chen, Y. TCP1 increases drug resistance in acute myeloid leukemia by suppressing autophagy via activating AKT/mTOR signaling. Cell Death Dis. 2021, 12, 1058. [Google Scholar] [CrossRef]
- Piano, V.; Alex, A.; Stege, P.; Maffini, S.; Stoppiello, G.A.; Veld, P.J.H.I.; Vetter, I.R.; Musacchio, A. CDC20 assists its catalytic incorporation in the mitotic checkpoint complex. Science 2021, 371, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Chang, L.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 2016, 536, 431–436. [Google Scholar] [CrossRef]
- Grantham, J.; Brackley, K.I.; Willison, K.R. Substantial CCT activity is required for cell cycle progression and cytoskeletal organization in mammalian cells. Exp. Cell Res. 2006, 312, 2309–2324. [Google Scholar] [CrossRef]
- Yokota, S.; Yanagi, H.; Yura, T.; Kubota, H. Cytosolic chaperonin-containing t-complex polypeptide 1 changes the content of a particular subunit species concomitant with substrate binding and folding activities during the cell cycle. Eur. J. Biochem. 2001, 268, 4664–4673. [Google Scholar] [CrossRef]
- Seixas, C.; Cruto, T.; Tavares, A.; Gaertig, J.; Soares, H. CCTalpha and CCTdelta chaperonin subunits are essential and required for cilia assembly and maintenance in Tetrahymena. PLoS ONE 2010, 5, e10704. [Google Scholar] [CrossRef]
- Kasembeli, M.; Lau, W.C.Y.; Roh, S.-H.; Eckols, T.K.; Frydman, J.; Chiu, W.; Tweardy, D.J. Modulation of STAT3 folding and function by TRiC/CCT chaperonin. PLoS Biol. 2014, 12, e1001844. [Google Scholar] [CrossRef]
- Kitamura, A.; Kubota, H.; Pack, C.-G.; Matsumoto, G.; Hirayama, S.; Takahashi, Y.; Kimura, H.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 2006, 8, 1163–1169. [Google Scholar] [CrossRef]
- Ghozlan, H.; Showalter, A.; Lee, E.; Zhu, X.; Khaled, A.R. Chaperonin-Containing TCP1 Complex (CCT) Promotes Breast Cancer Growth Through Correlations with Key Cell Cycle Regulators. Front. Oncol. 2021, 11, 663877. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Jiao, S.; Zhou, Z. STRIPAK complexes in cell signaling and cancer. Oncogene 2016, 35, 4549–4557. [Google Scholar] [CrossRef]
- Tang, N.; Cai, X.; Peng, L.; Liu, H.; Chen, Y. TCP1 regulates Wnt7b/beta-catenin pathway through P53 to influence the proliferation and migration of hepatocellular carcinoma cells. Signal Transduct. Target. Ther. 2020, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Roh, S.-H.; Kasembeli, M.M.; Galaz-Montoya, J.G.; Chiu, W.; Tweardy, D.J. Chaperonin TRiC/CCT Recognizes Fusion Oncoprotein AML1-ETO through Subunit-Specific Interactions. Biophys. J. 2016, 110, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Roh, S.-H.; Kasembeli, M.; Galaz-Montoya, J.G.; Trnka, M.; Lau, W.C.-Y.; Burlingame, A.; Chiu, W.; Tweardy, D.J. Chaperonin TRiC/CCT Modulates the Folding and Activity of Leukemogenic Fusion Oncoprotein AML1-ETO. J. Biol. Chem. 2016, 291, 4732–4741. [Google Scholar] [CrossRef]
- Guo, X.-D.; Ji, J.; Xue, T.-F.; Sun, Y.-Q.; Guo, R.-B.; Cheng, H.; Sun, X.-L. FTY720 Exerts Anti-Glioma Effects by Regulating the Glioma Microenvironment Through Increased CXCR4 Internalization by Glioma-Associated Microglia. Front. Immunol. 2020, 11, 178. [Google Scholar] [CrossRef]
- Davy, M.; Genest, L.; Legrand, C.; Pelouin, O.; Froget, G.; Castagné, V.; Rupp, T. Evaluation of Temozolomide and Fingolimod Treatments in Glioblastoma Preclinical Models. Cancers 2023, 15, 4478. [Google Scholar] [CrossRef]
- Hirata, N.; Yamada, S.; Yanagida, S.; Ono, A.; Kanda, Y. FTY720 Inhibits Expansion of Breast Cancer Stem Cells via PP2A Activation. Int. J. Mol. Sci. 2021, 22, 7259. [Google Scholar] [CrossRef]
- Laroche, F.J.F.; Li, S.; Shen, N.; Hwang, S.K.; Nguyen, G.; Yu, W.; Wong, C.K.; Quinton, R.J.; Berman, J.N.; Liu, C.-T.; et al. S1P1 Threonine 236 Phosphorylation Mediates the Invasiveness of Triple-Negative Breast Cancer and Sensitivity to FTY720. Cells 2023, 12, 980. [Google Scholar] [CrossRef]
- Lee, J.; Ryu, J.; Yoon, G.; Jeon, H.; Cho, Y.; Choi, J.; Song, S.Y.; Do, I.; Lee, Y.; Kim, T.; et al. Sphingosine kinase 1 as a potential therapeutic target in epithelial ovarian cancer. Int. J. Cancer 2015, 137, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Kreitzburg, K.M.; Fehling, S.C.; Landen, C.N.; Gamblin, T.L.; Vance, R.B.; Arend, R.C.; Katre, A.A.; Oliver, P.G.; van Waardenburg, R.C.; Alvarez, R.D.; et al. FTY720 enhances the anti-tumor activity of carboplatin and tamoxifen in a patient-derived xenograft model of ovarian cancer. Cancer Lett. 2018, 436, 75–86. [Google Scholar] [CrossRef]
- Young, M.M.; Bui, V.; Chen, C.; Wang, H.-G. FTY720 induces non-canonical phosphatidylserine externalization and cell death in acute myeloid leukemia. Cell Death Dis. 2019, 10, 847. [Google Scholar] [CrossRef] [PubMed]
- Vorbach, S.; Gründer, A.; Zhou, F.; Koellerer, C.; Jutzi, J.S.; Simoni, M.; Riccetti, L.; Valk, P.J.; Sanders, M.A.; Müller-Tidow, C.; et al. Enhanced expression of the sphingosine-1-phosphate-receptor-3 causes acute myelogenous leukemia in mice. Leukemia 2020, 34, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Luo, L.-F.; Lu, J.; Li, L.; Liu, Y.-F.; Wang, J.; Liu, H.; Song, H.; Jiang, H.; Chen, S.-J.; et al. FTY720 induces apoptosis of M2 subtype acute myeloid leukemia cells by targeting sphingolipid metabolism and increasing endogenous ceramide levels. PLoS ONE 2014, 9, e103033. [Google Scholar] [CrossRef]
- Arriazu, E.; Vicente, C.; Pippa, R.; Peris, I.; Martínez-Balsalobre, E.; García-Ramírez, P.; Marcotegui, N.; Igea, A.; Alignani, D.; Rifón, J.; et al. A new regulatory mechanism of protein phosphatase 2A activity via SET in acute myeloid leukemia. Blood Cancer J. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Thol, F.; Ganser, A. Treatment of Relapsed Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2020, 21, 66. [Google Scholar] [CrossRef]
- Chen, J.-H.; Chen, Y.Z.; Wu, Y. Establishment and characterization of highly tumorigenic leukemia cell line HL-60 transplanted through repeated passages into nude mice. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2010, 18, 350–354. [Google Scholar]
- Newell, L.F.; Cook, R.J. Advances in acute myeloid leukemia. BMJ 2021, 375, n2026. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, X.; Tian, X. High expression of long intergenic non-coding RNA LINC00662 contributes to malignant growth of acute myeloid leukemia cells by upregulating ROCK1 via sponging microRNA-340-5p. Eur. J. Pharmacol. 2019, 859, 172535. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Z.; Lv, L.; Li, P.; Xiu, B.; Qian, W.; Liang, A. MiRNA-340-5p mediates the functional and infiltrative promotion of tumor-infiltrating CD8(+) T lymphocytes in human diffuse large B cell lymphoma. J. Exp. Clin. Cancer Res. 2020, 39, 238. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Rong, A.; Zhou, Q.; Li, W. LncRNA LINC00662 promotes colon cancer tumor growth and metastasis by competitively binding with miR-340-5p to regulate CLDN8/IL22 co-expression and activating ERK signaling pathway. J. Exp. Clin. Cancer Res. 2020, 39, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, X.; Lu, B.; Gu, Y.; Chen, Q.; Lei, T.; Nie, F.; Gu, J.; Huang, J.; Wei, C.; et al. Up-regulated LINC01234 promotes non-small-cell lung cancer cell metastasis by activating VAV3 and repressing BTG2 expression. J. Hematol. Oncol. 2020, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.; Zhang, Y.; Wang, H.; Rong, X.; Peng, J.; He, L.; Peng, Y. An miR-340-5p-macrophage feedback loop modulates the progression and tumor microenvironment of glioblastoma multiforme. Oncogene 2019, 38, 7399–7415. [Google Scholar] [CrossRef]
- Vicente, C.; Arriazu, E.; Martínez-Balsalobre, E.; Peris, I.; Marcotegui, N.; García-Ramírez, P.; Pippa, R.; Rabal, O.; Oyarzábal, J.; Guruceaga, E.; et al. A novel FTY720 analogue targets SET-PP2A interaction and inhibits growth of acute myeloid leukemia cells without inducing cardiac toxicity. Cancer Lett. 2020, 468, 1–13. [Google Scholar] [CrossRef]
- McCracken, A.N.; McMonigle, R.J.; Tessier, J.; Fransson, R.; Perryman, M.S.; Chen, B.; Keebaugh, A.; Selwan, E.; Barr, S.A.; Kim, S.M.; et al. Phosphorylation of a constrained azacyclic FTY720 analog enhances anti-leukemic activity without inducing S1P receptor activation. Leukemia 2017, 31, 669–677. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Tu, G.; Yuan, Y.; Liu, J.; Jiang, Q.; Liu, Y.; Wu, Q.; Wu, L.; Chen, Y. The Molecular Chaperone TCP1 Affects Carcinogenicity and Is a Potential Therapeutic Target for Acute Myeloid Leukemia. Pharmaceutics 2025, 17, 557. https://doi.org/10.3390/pharmaceutics17050557
Wu Y, Tu G, Yuan Y, Liu J, Jiang Q, Liu Y, Wu Q, Wu L, Chen Y. The Molecular Chaperone TCP1 Affects Carcinogenicity and Is a Potential Therapeutic Target for Acute Myeloid Leukemia. Pharmaceutics. 2025; 17(5):557. https://doi.org/10.3390/pharmaceutics17050557
Chicago/Turabian StyleWu, Yong, Guihui Tu, Yuxia Yuan, Jingwen Liu, Qingna Jiang, Yang Liu, Qiurong Wu, Lixian Wu, and Yuanzhong Chen. 2025. "The Molecular Chaperone TCP1 Affects Carcinogenicity and Is a Potential Therapeutic Target for Acute Myeloid Leukemia" Pharmaceutics 17, no. 5: 557. https://doi.org/10.3390/pharmaceutics17050557
APA StyleWu, Y., Tu, G., Yuan, Y., Liu, J., Jiang, Q., Liu, Y., Wu, Q., Wu, L., & Chen, Y. (2025). The Molecular Chaperone TCP1 Affects Carcinogenicity and Is a Potential Therapeutic Target for Acute Myeloid Leukemia. Pharmaceutics, 17(5), 557. https://doi.org/10.3390/pharmaceutics17050557