
A Convenient Strategy for Studying Antibody Aggregation and Inhibition of Aggregation: Characterization and Simulation

Abstract

1. Introduction
2. Materials and Methods
2.1. Thermal Incubation Experiments
2.2. UV Absorption
2.3. SEC-HPLC
2.4. Dynamic Light Scattering
2.5. Circular Dichroism
2.6. Molecular Dynamics Simulations
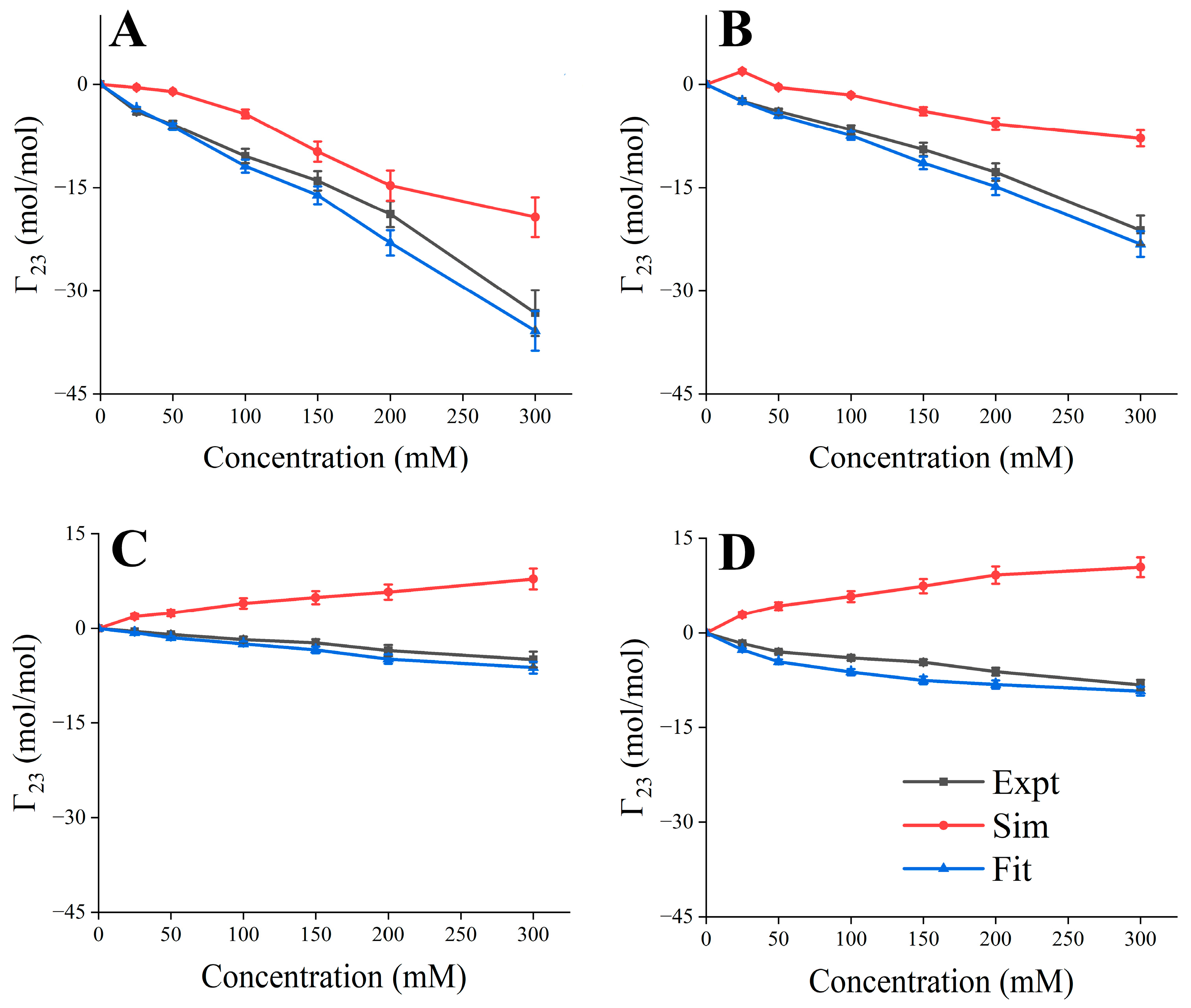
2.7. Experimental and Simulated Preferential Interaction Coefficient
3. Results and Discussion
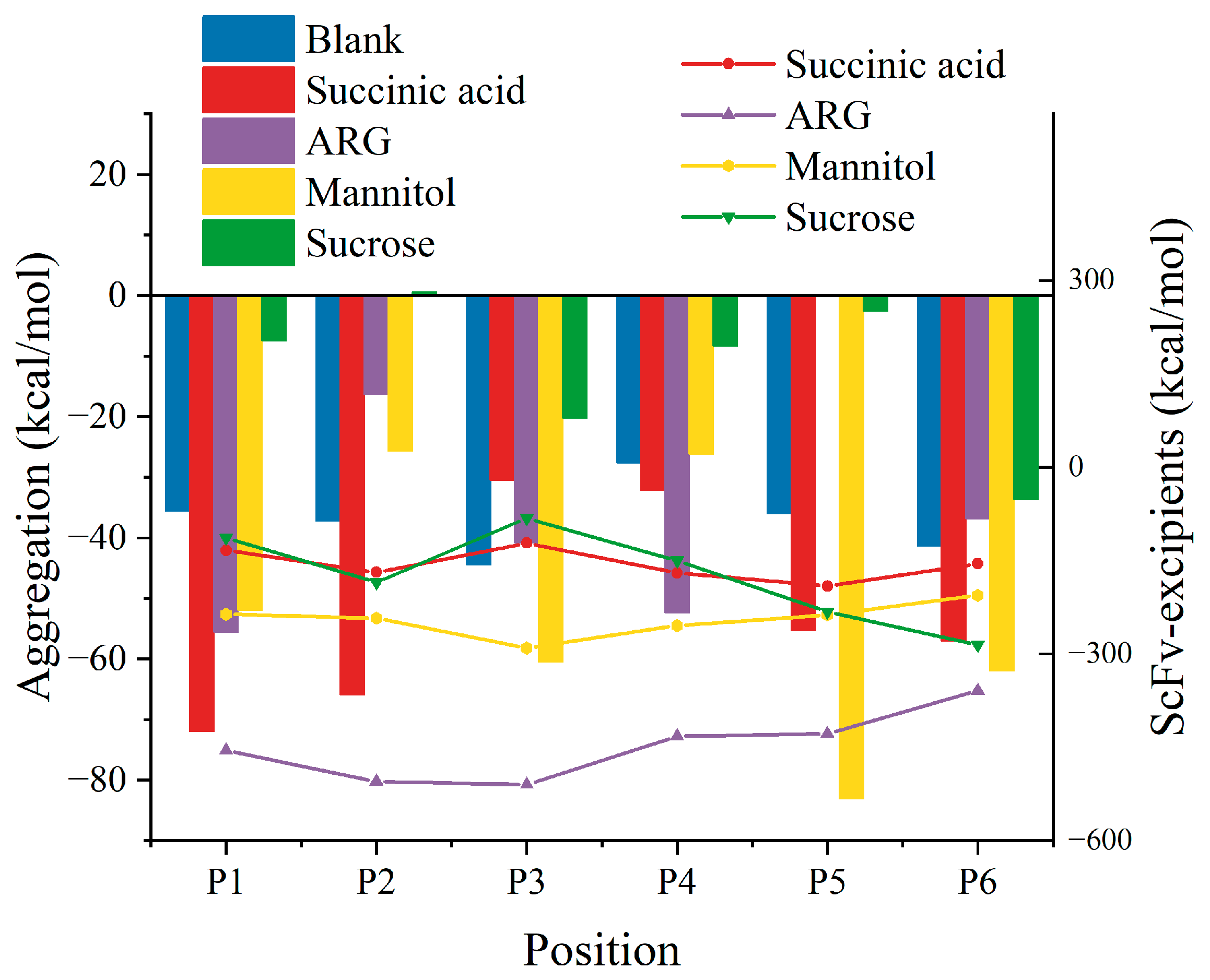
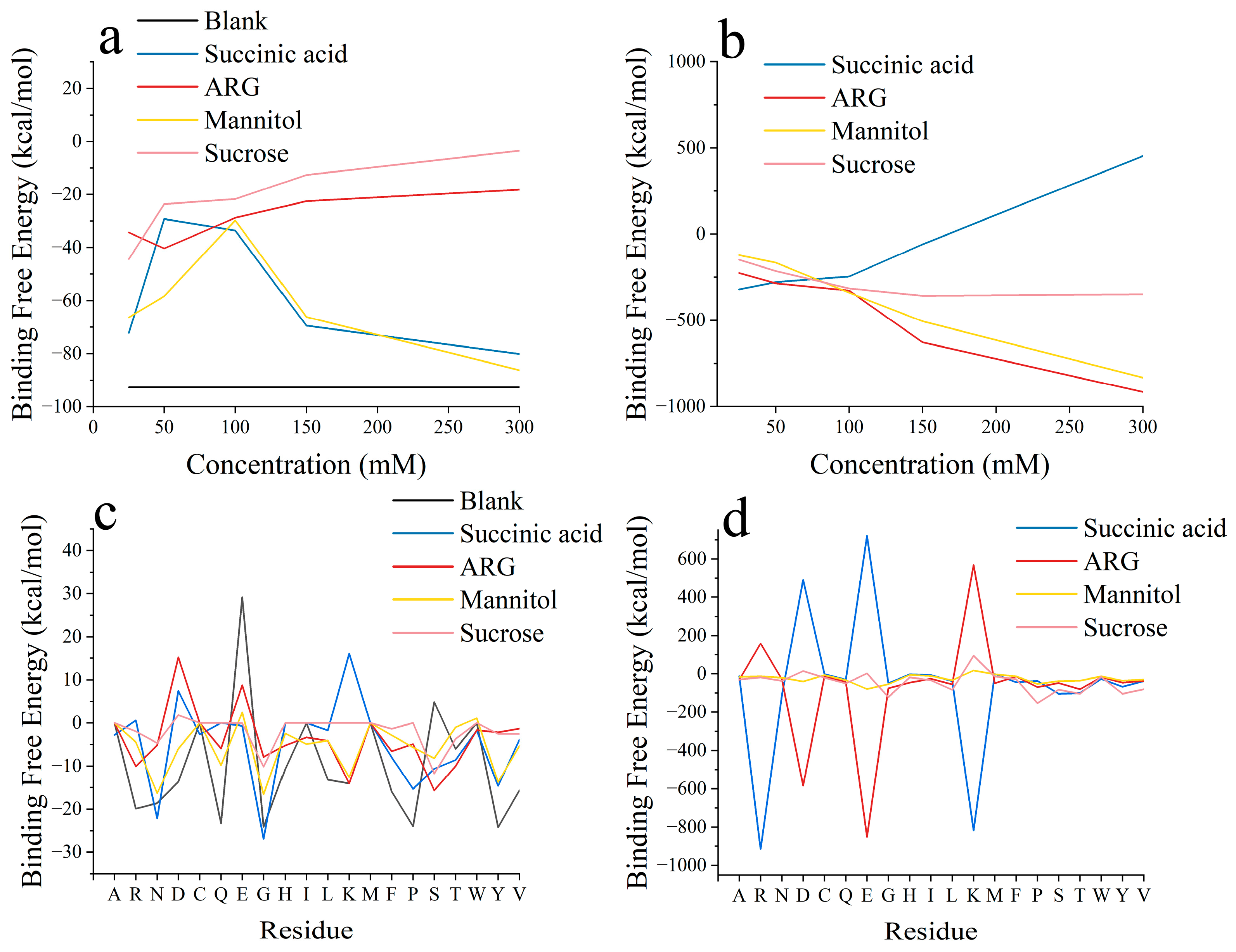
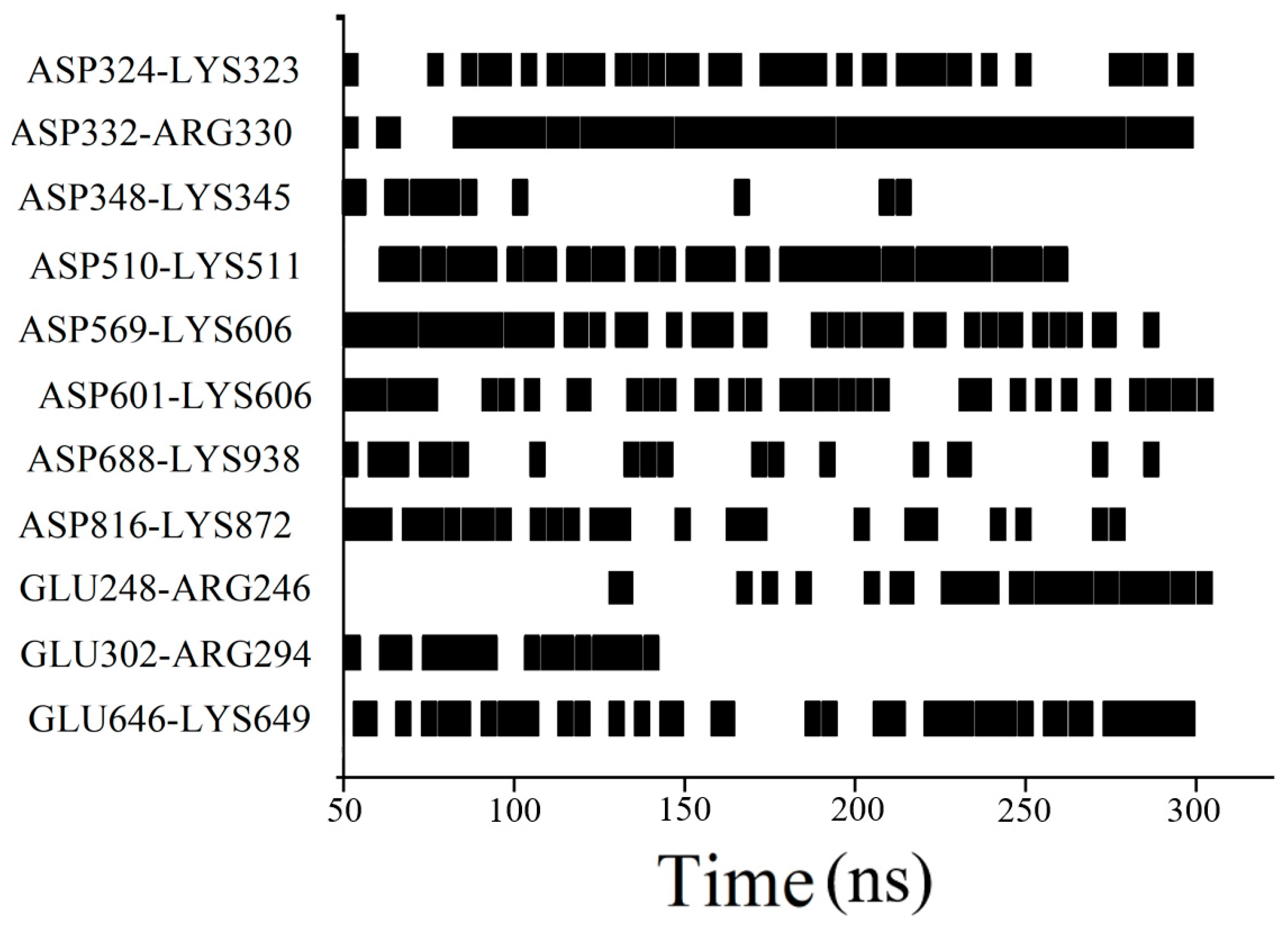
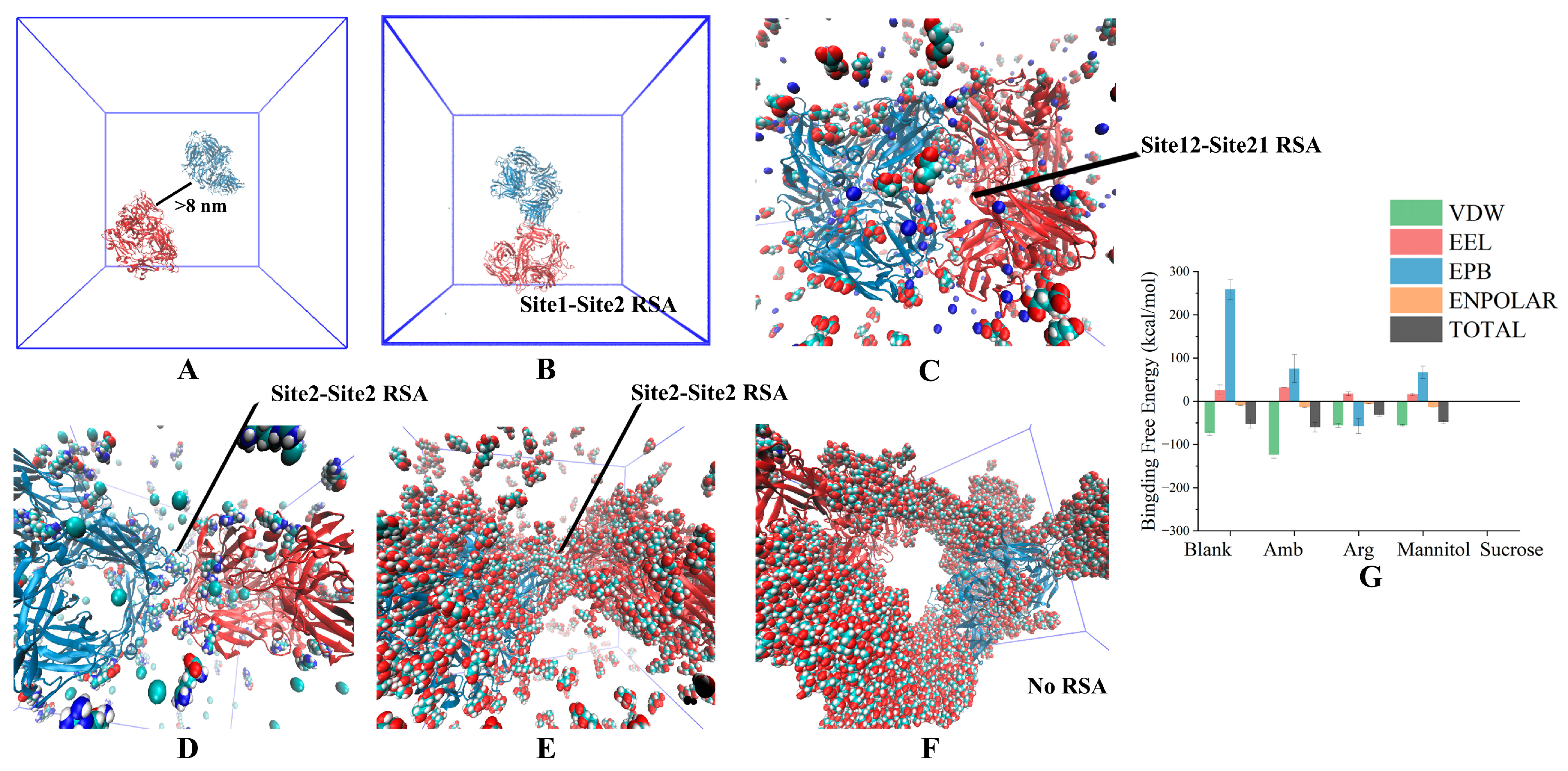
3.1. Two-Molecule Simulations with Different Initial Positions
3.2. Excipient Concentration Simulation
3.3. Long-Time Annealing Simulation
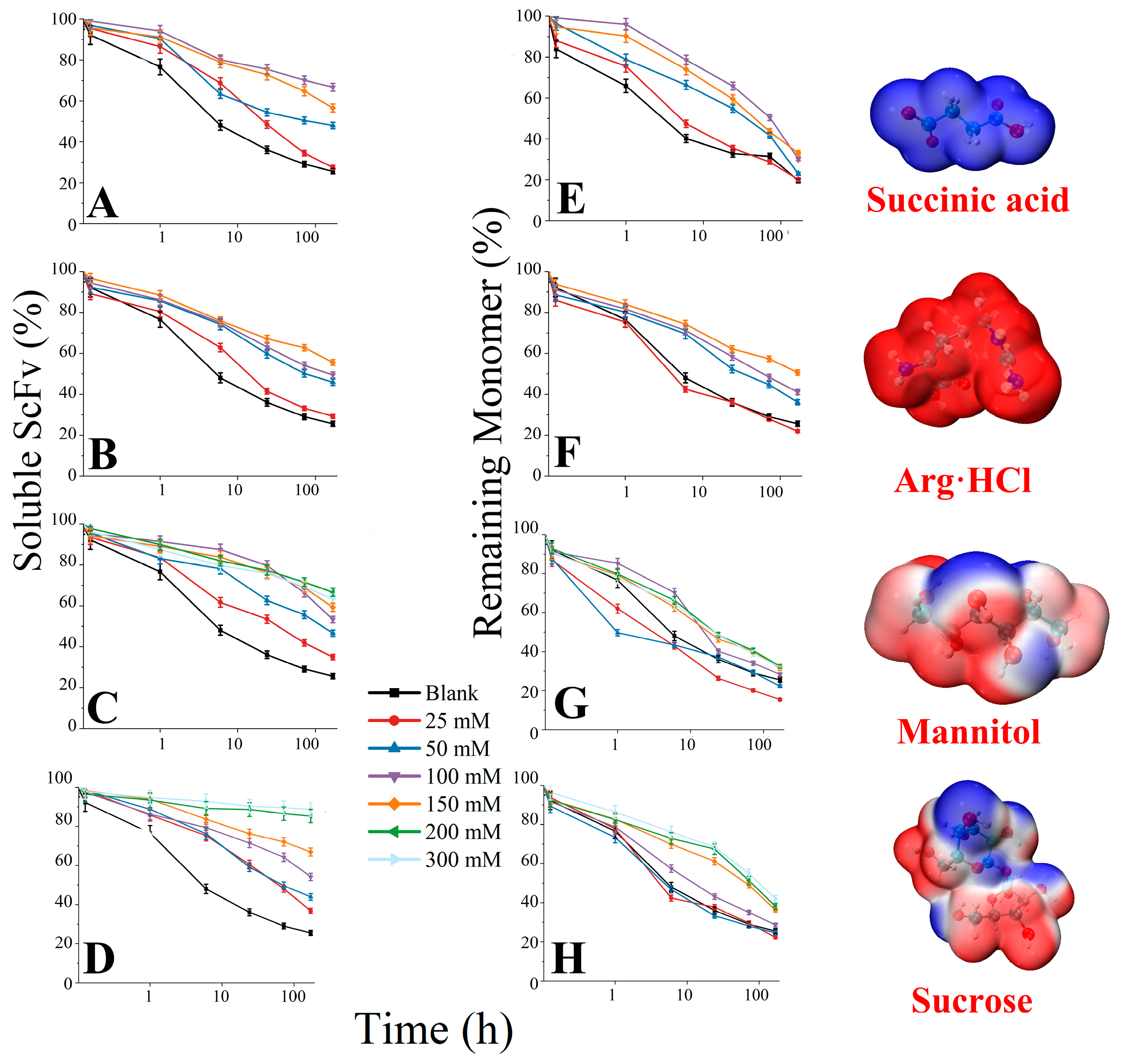
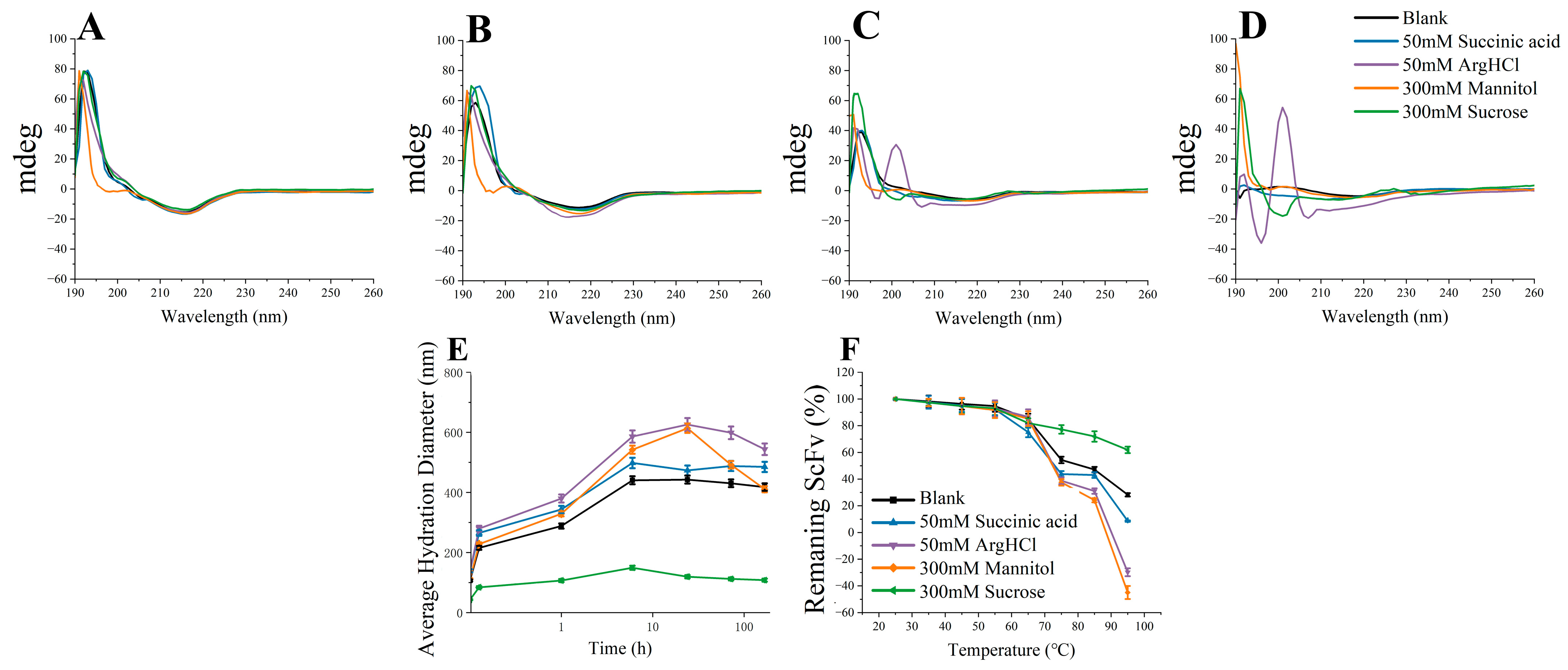
3.4. Soluble BsScFv Characterization
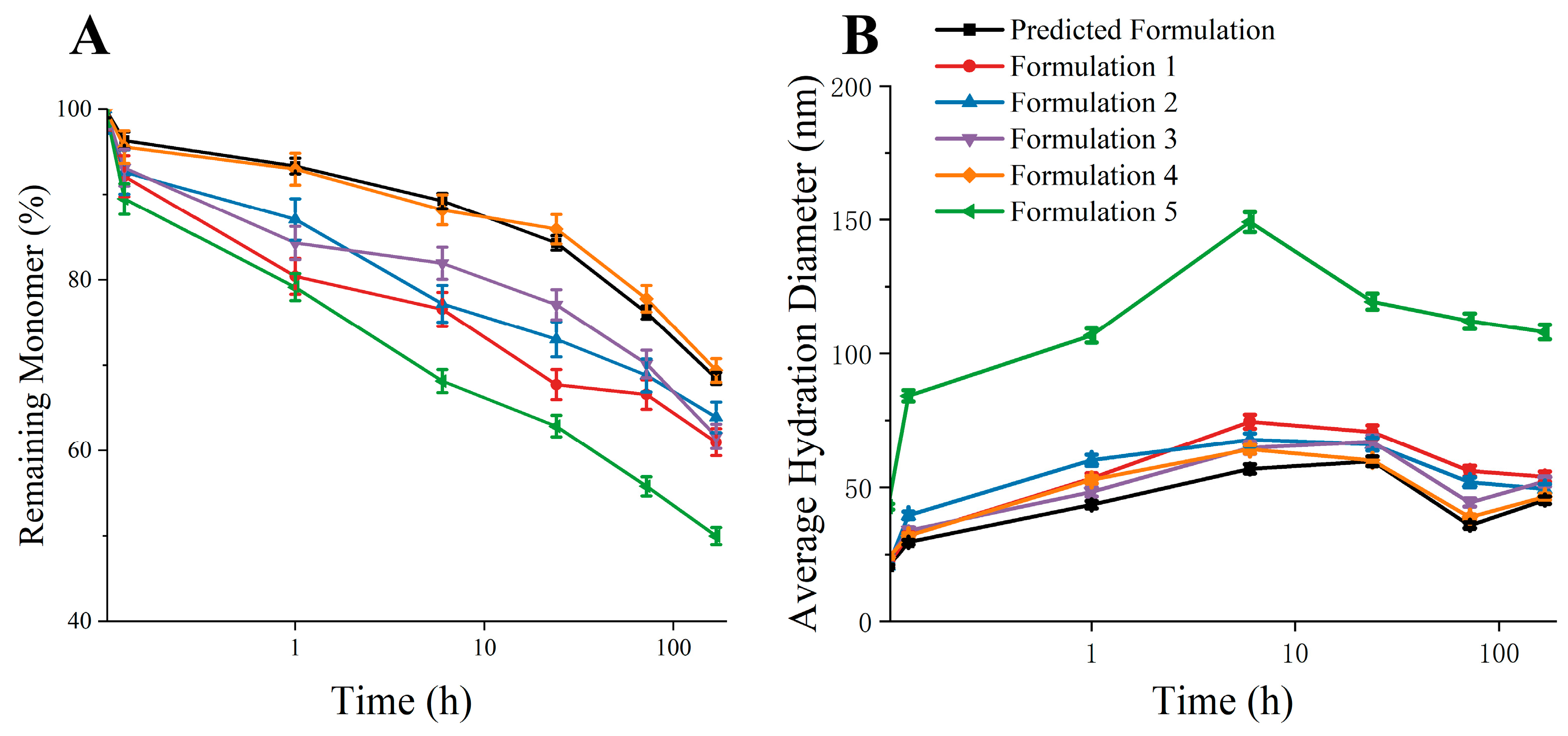
3.5. Validation of Predicted Formulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, Y.; Yu, X.; Wang, X.; Yuan, K.; Wang, G.; Hu, L.; Zhang, G.; Pei, W.; Wang, L.; Sun, C.; et al. Bispecific antibodies in cancer therapy: Target selection and regulatory requirements. Acta Pharm. Sin. B 2023, 13, 3583–3597. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Jiang, M.; Li, T.; Zhu, J.; Liu, K.; Hou, H.; Zhang, X. Anlotinib combined with anti-PD-1 antibody, camrelizumab for advanced NSCLCs after multiple lines treatment: An open-label, dose escalation and expansion study. Lung Cancer 2021, 160, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hempel, P.; Muller, P.; Oruzio, D.; Behr, W.; Brockmeyer, C.; Wochner, M.; Ehnle, S.; Riethmuller, R.; Schlimok, G. Combination of high-dose chemotherapy and monoclonal antibody in breast-cancer patients: A pilot trial to monitor treatment effects on disseminated tumor cells. Cytotherapy 2000, 2, 287–295. [Google Scholar] [CrossRef]
- Kollar, E.; Balazs, B.; Tari, T.; Siro, I. Development challenges of high concentration monoclonal antibody formulations. Drug Discov. Today Technol. 2020, 37, 31–40. [Google Scholar] [CrossRef]
- Roberts, C.J. Protein aggregation and its impact on product quality. Curr. Opin. Biotechnol. 2014, 30, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Chou, D.K.; Jiskoot, W.; Arosio, P. Nucleation in Protein Aggregation in Biotherapeutic Development: A look into the Heart of the Event. J. Pharm. Sci. 2022, 111, 951–959. [Google Scholar] [CrossRef]
- Le Basle, Y.; Chennell, P.; Tokhadze, N.; Astier, A.; Sautou, V. Physicochemical Stability of Monoclonal Antibodies: A Review. J. Pharm. Sci. 2020, 109, 169–190. [Google Scholar] [CrossRef]
- Austerberry, J.I.; Dajani, R.; Panova, S.; Roberts, D.; Golovanov, A.P.; Pluen, A.; van der Walle, C.F.; Uddin, S.; Warwicker, J.; Derrick, J.P.; et al. The effect of charge mutations on the stability and aggregation of a human single chain Fv fragment. Eur. J. Pharm. Biopharm. 2017, 115, 18–30. [Google Scholar] [CrossRef]
- Arya, A.; Rathee, J.; Kishore, N. Analyzing synergy in combined influence of sorbitol and glycine betaine on protein Stability: Thermodynamic and mechanistic insights. J. Mol. Liq. 2024, 400, 124515. [Google Scholar] [CrossRef]
- Wu, X.; Deng, F.; Chen, Y.; Xu, M.; Ma, F.; Shi, L. Electrostatic and hydrophobic interaction cooperative nanochaperone regulates protein folding. Aggregate 2023, 5, e429. [Google Scholar] [CrossRef]
- Paul, R.; Bera, S.; Devi, M.; Paul, S. Inhibition of Aβ16–22 Peptide Aggregation by Small Molecules and Their Permeation through POPC Lipid Bilayer: Insight from Molecular Dynamics Simulation Study. J. Chem. Inf. Model. 2022, 62, 5193–5207. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, Z.; Gierasch, L.M. Inhibition of protein aggregation in vitro and in vivo by a natural osmoprotectant. Proc. Natl. Acad. Sci. USA 2006, 103, 13357–13361. [Google Scholar] [CrossRef] [PubMed]
- Lao, Z.; Chen, Y.; Tang, Y.; Wei, G. Molecular Dynamics Simulations Reveal the Inhibitory Mechanism of Dopamine against Human Islet Amyloid Polypeptide (hIAPP) Aggregation and Its Destabilization Effect on hIAPP Protofibrils. ACS Chem. Neurosci. 2019, 10, 4151–4159. [Google Scholar] [CrossRef]
- Ye, Y.; Huo, X.; Yin, Z. Protein-protein interactions at high concentrations: Effects of ArgHCl and NaCl on the stability, viscosity and aggregation mechanisms of protein solution. Int. J. Pharm. 2021, 601, 120535. [Google Scholar] [CrossRef] [PubMed]
- Austerberry, J.I.; Thistlethwaite, A.; Fisher, K.; Golovanov, A.P.; Pluen, A.; Esfandiary, R.; van der Walle, C.F.; Warwicker, J.; Derrick, J.P.; Curtis, R. Arginine to Lysine Mutations Increase the Aggregation Stability of a Single-Chain Variable Fragment through Unfolded-State Interactions. Biochemistry 2019, 58, 3413–3421. [Google Scholar] [CrossRef]
- Ng, Y.K.; Konermann, L. Mechanism of Protein Aggregation Inhibition by Arginine: Blockage of Anionic Side Chains Favors Unproductive Encounter Complexes. J. Am. Chem. Soc. 2024, 146, 8394–8406. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Li, R.; Xiang, D. L-lysine moderates thermal aggregation of coconut proteins induced by thermal treatment. Sci. Rep. 2023, 13, 13310. [Google Scholar] [CrossRef]
- Pyne, P.; Pyne, S.; Mitra, R.K. Sugar Molecules Inhibit Insulin Aggregation: A Decisive Role Being Played by the Protein Solvation Energetics. J. Phys. Chem. B 2023, 127, 8825–8832. [Google Scholar] [CrossRef]
- Pradhan, N.; Shekhar, S.; Jana, N.R.; Jana, N.R. Sugar-Terminated Nanoparticle Chaperones Are 102–105 Times Better Than Molecular Sugars in Inhibiting Protein Aggregation and Reducing Amyloidogenic Cytotoxicity. ACS Appl. Mater. Interfaces 2017, 9, 10554–10566. [Google Scholar] [CrossRef]
- Blanco, M.A. Computational models for studying physical instabilities in high concentration biotherapeutic formulations. mAbs 2022, 14, 2044744. [Google Scholar] [CrossRef]
- Meric, G.; Robinson, A.S.; Roberts, C.J. Driving Forces for Nonnative Protein Aggregation and Approaches to Predict Aggregation-Prone Regions. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Saikia, B.; Gogoi, C.R.; Baruah, A. Advances in the understanding of protein misfolding and aggregation through molecular dynamics simulation. Prog. Biophys. Mol. Biol. 2022, 175, 31–48. [Google Scholar] [CrossRef]
- Planas-Iglesias, J.; Borko, S.; Swiatkowski, J.; Elias, M.; Havlasek, M.; Salamon, O.; Grakova, E.; Kunka, A.; Martinovic, T.; Damborsky, J.; et al. AggreProt: A web server for predicting and engineering aggregation prone regions in proteins. Nucleic Acids Res. 2024, 52, W159–W169. [Google Scholar] [CrossRef]
- Tartaglia, G.G.; Vendruscolo, M. The Zyggregator method for predicting protein aggregation propensities. Chem. Soc. Rev. 2008, 37, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Rojas, G.; Relova-Hernández, E.; Pérez-Riverón, A.; Castro-Martínez, C.; Diaz-Bravo, O.; Infante, Y.C.; Gómez, T.; Solozábal, J.; DíazBravo, A.B.; Schubert, M.; et al. Molecular reshaping of phage-displayed Interleukin-2 at beta chain receptor interface to obtain potent super-agonists with improved developability profiles. Commun. Biol. 2023, 6, 828. [Google Scholar] [CrossRef]
- Baumketner, A.; Shea, J.-E. The Structure of the Alzheimer Amyloid β 10-35 Peptide Probed through Replica-Exchange Molecular Dynamics Simulations in Explicit Solvent. J. Mol. Biol. 2007, 366, 275–285. [Google Scholar] [CrossRef]
- Uchiyama, S. Liquid formulation for antibody drugs. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2014, 1844, 2041–2052. [Google Scholar] [CrossRef]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Izadi, S. Water models for biomolecular simulations. WIREs Comput. Mol. Sci. 2017, 8, e1347. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- London, N.; Ambroggio, X. An accurate binding interaction model in de novo computational protein design of interactions: If you build it, they will bind. J. Struct. Biol. 2014, 185, 136–146. [Google Scholar] [CrossRef]
- Li, W.; Zhang, J.; Su, Y.; Wang, J.; Qin, M.; Wang, W. Effects of Zinc Binding on the Conformational Distribution of the Amyloid-β Peptide Based on Molecular Dynamics Simulations. J. Phys. Chem. B 2007, 111, 13814–13821. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Myers, J.; Grothaus, G.; Narayanan, S.; Onufriev, A. A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins Struct. Funct. Bioinform. 2006, 63, 928–938. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibsom, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Schauperl, M.; Nerenberg, P.S.; Jang, H.; Wang, L.-P.; Bayly, C.I.; Mobley, D.L.; Gilson, M.K. Non-bonded force field model with advanced restrained electrostatic potential charges (RESP2). Commun. Chem. 2020, 3, 44. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Hong, J.; Capp, M.W.; Anderson, C.F.; Saecker, R.M.; Felitsky, D.J.; Anderson, M.W.; Record, M.T. Preferential Interactions of Glycine Betaine and of Urea with DNA: Implications for DNA Hydration and for Effects of These Solutes on DNA Stability. Biochemistry 2004, 43, 14744–14758. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Shinde, C.; Trout, B.L. Molecular Computations of Preferential Interaction Coefficients of Proteins. J. Phys. Chem. B 2009, 113, 12546–12554. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, T.K.; Sudrik, C.; Mody, N.; Hasige, S.A.; Trout, B.L. Molecular computations of preferential interactions of proline, arginine. HCl, and NaCl with IgG1 antibodies and their impact on aggregation and viscosity. MAbs 2020, 12, 1816312. [Google Scholar] [CrossRef]
- Cloutier, T.; Sudrik, C.; Sathish, H.A.; Trout, B.L. Kirkwood–Buff-Derived Alcohol Parameters for Aqueous Carbohydrates and Their Application to Preferential Interaction Coefficient Calculations of Proteins. J. Phys. Chem. B 2018, 122, 9350–9360. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Formulation |
|---|---|
| Predicted Formulation | 50 mM succinic acid + 150 mM Arg·HCl + 200 mM Sucrose |
| Formulation 1 | 15 mM succinic acid + 50 mM Arg·HCl + 50 mM Sucrose |
| Formulation 2 | 30 mM succinic acid + 100 mM Arg·HCl + 100 mM Sucrose |
| Formulation 3 | 75 mM succinic acid + 200 mM Arg·HCl + 150 mM Sucrose |
| Formulation 4 | 100 mM succinic acid + 300 mM Arg·HCl + 300 mM Sucrose |
| Formulation 5 | 50 mM succinic acid + 150 mM Arg·HCl + 200 mM Sucrose + 100 mM Mannitol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Chen, X.; Fang, G.; Cao, X.; Wan, J. A Convenient Strategy for Studying Antibody Aggregation and Inhibition of Aggregation: Characterization and Simulation. Pharmaceutics 2025, 17, 534. https://doi.org/10.3390/pharmaceutics17040534
Guo Y, Chen X, Fang G, Cao X, Wan J. A Convenient Strategy for Studying Antibody Aggregation and Inhibition of Aggregation: Characterization and Simulation. Pharmaceutics. 2025; 17(4):534. https://doi.org/10.3390/pharmaceutics17040534
Chicago/Turabian StyleGuo, Yibo, Xi Chen, Guchen Fang, Xuejun Cao, and Junfen Wan. 2025. "A Convenient Strategy for Studying Antibody Aggregation and Inhibition of Aggregation: Characterization and Simulation" Pharmaceutics 17, no. 4: 534. https://doi.org/10.3390/pharmaceutics17040534
APA StyleGuo, Y., Chen, X., Fang, G., Cao, X., & Wan, J. (2025). A Convenient Strategy for Studying Antibody Aggregation and Inhibition of Aggregation: Characterization and Simulation. Pharmaceutics, 17(4), 534. https://doi.org/10.3390/pharmaceutics17040534