
Development of Multifunctional Targeted Dual-Loaded Polymeric Nanoparticles for Triple-Negative Breast Cancer Treatment


Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
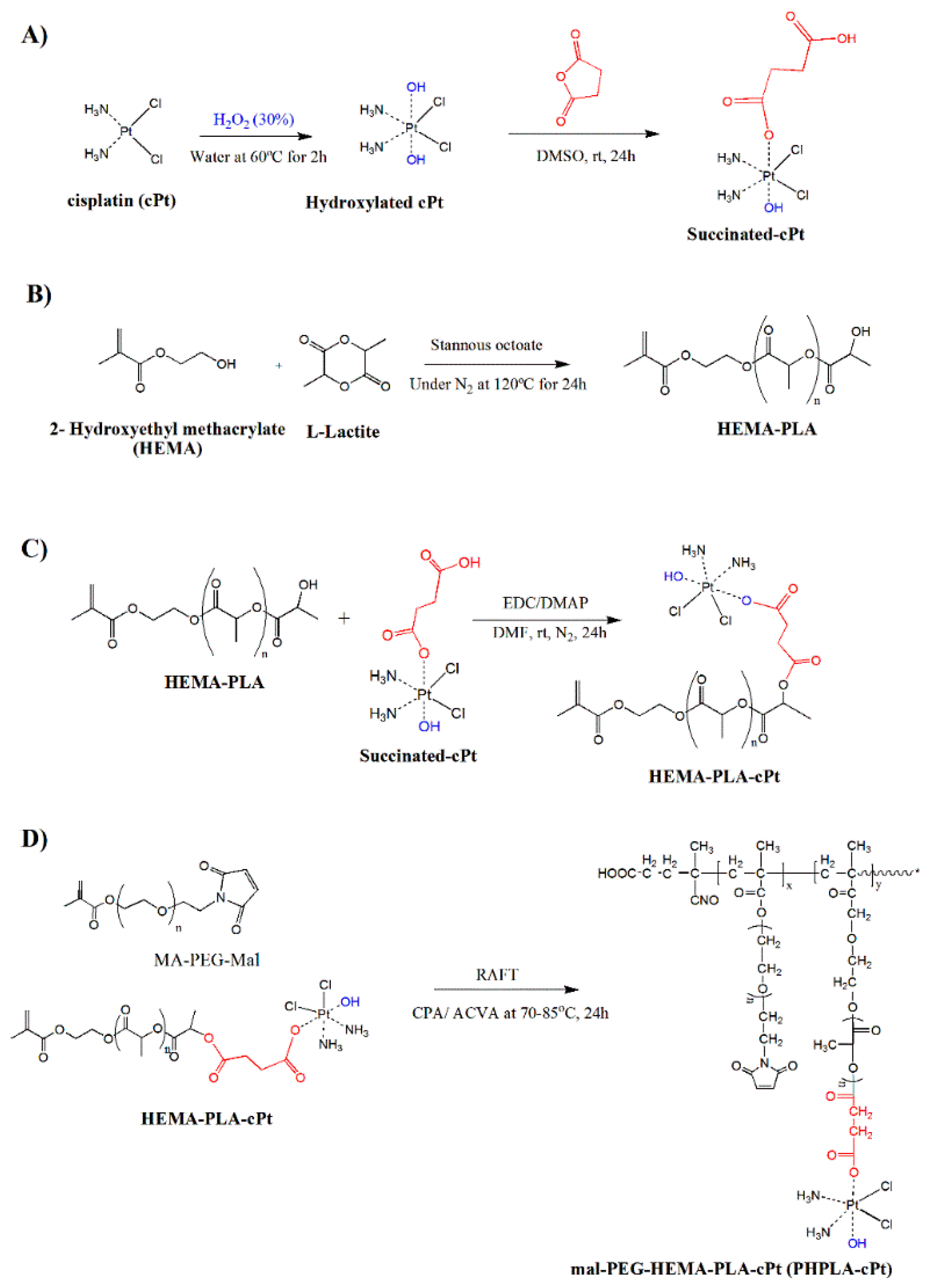
2.2. Oxidation of Cisplatin (cPt)
2.3. Synthesis of Oxidized Cisplatin-Succinate Conjugate
2.4. Synthesis of HEMA-Poly (L(-)-Lactide Macromonomer (HEMA-PLA)
2.5. Synthesis of HEMA-PLA Macromonomer-Cisplatin Conjugate (HPLA-cPt)
2.6. Synthesis of Poly (Mal-Peg-HPLA-cPt) by RAFT (Reversible Addition Fragmentation Chain Transfer) Polymerization
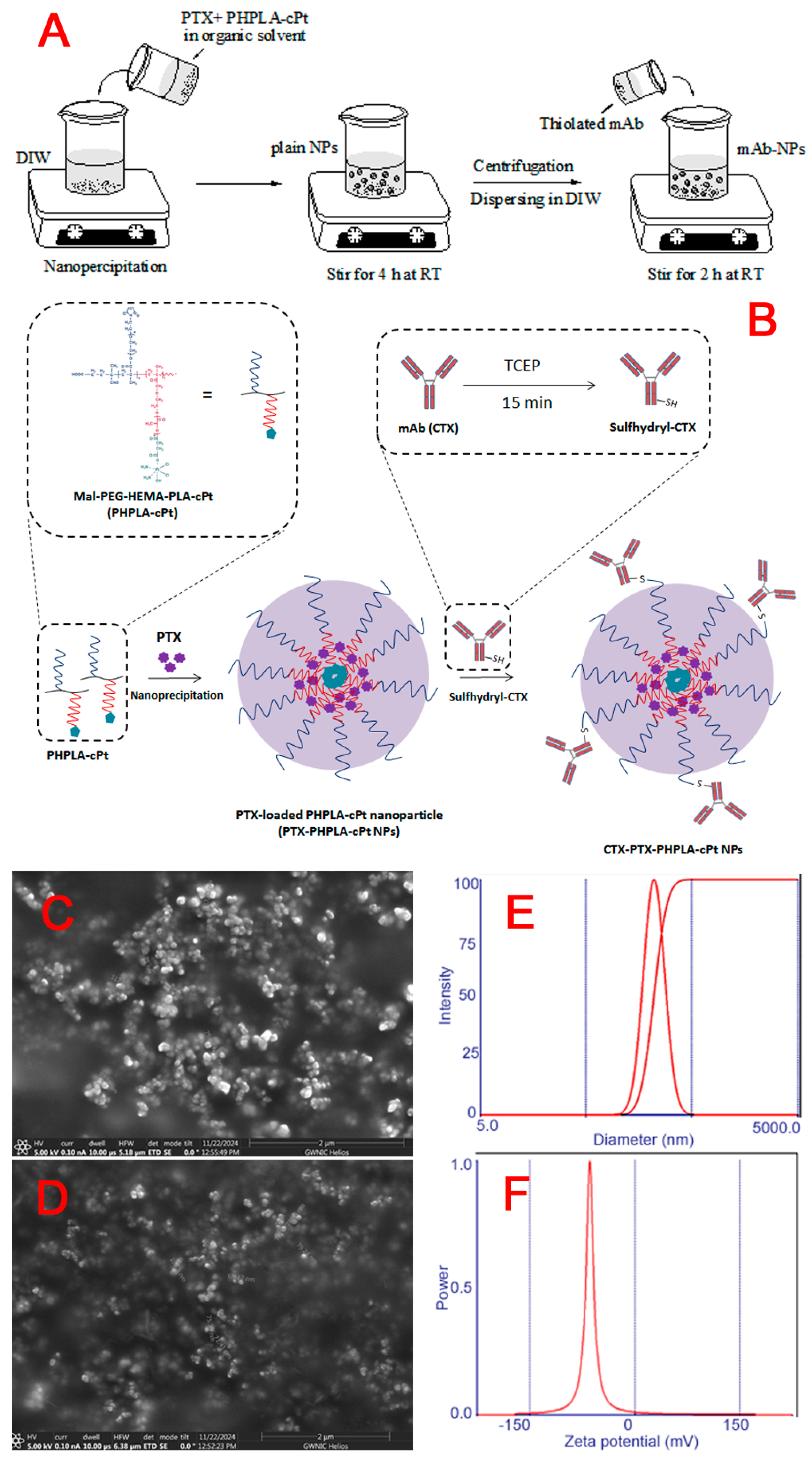
2.7. Preparation of PTX-Loaded Poly (Mal-Peg- HPLA-cPt) Nanoparticles (PTX-PHPLA-cPt-NPs)
2.8. Conjugation of Antibody to the Nanoparticle via Thiol-Maleimide Chemistry
2.9. Measurement of Hydrodynamic Size, Zeta Potential, and Morphology
2.10. In Vitro Drug Availability Study
2.11. In Vitro Antiproliferation Study
2.12. In Vitro Cellular Uptake
2.13. Intracellular Localization of Nanoparticles by Confocal Microscopy
2.14. Cell Binding Assay of CTX-Nanoparticles by Flow Cytometry Analysis
2.15. Statistical Analysis
3. Results
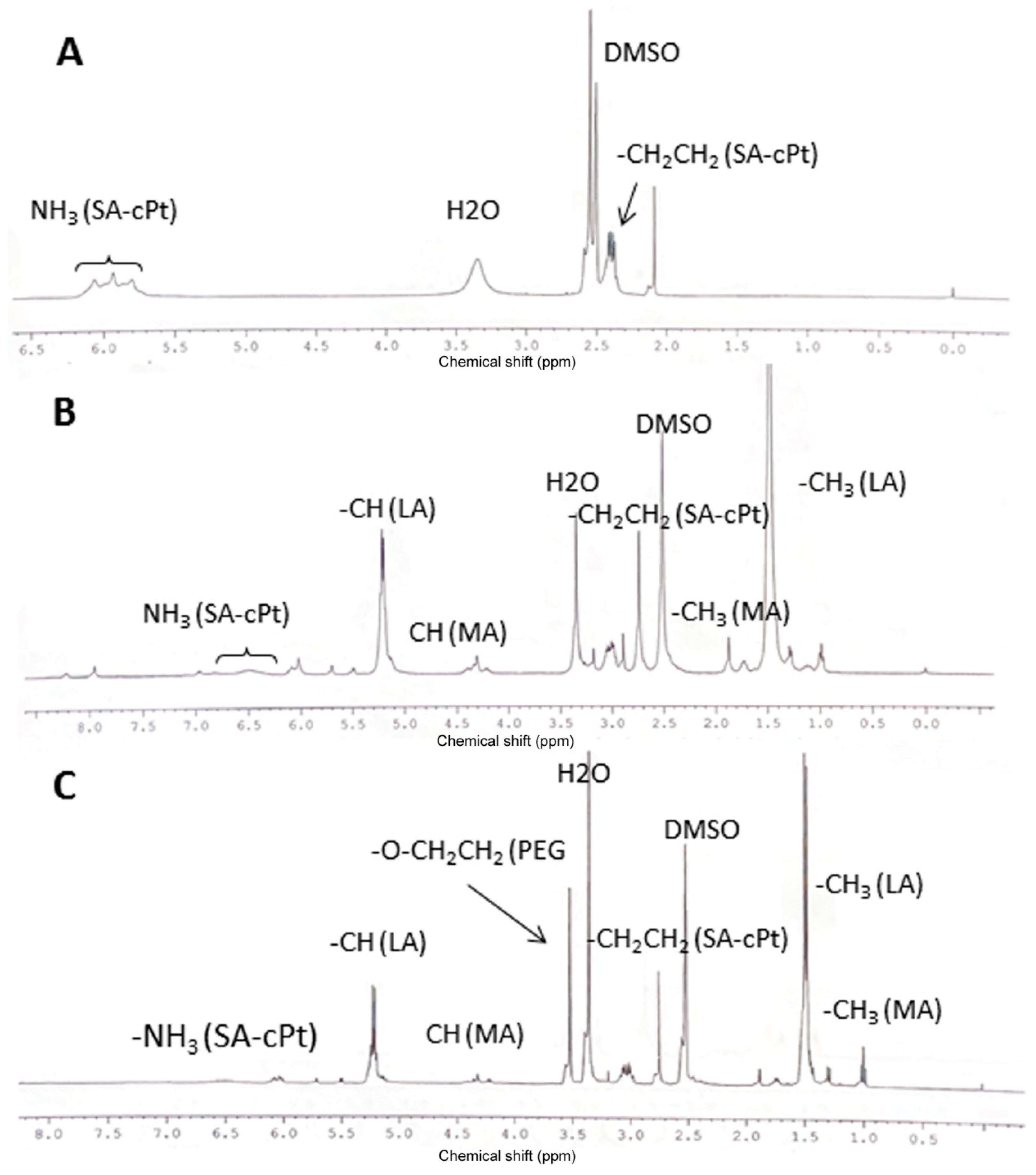
3.1. Cisplatin Modification
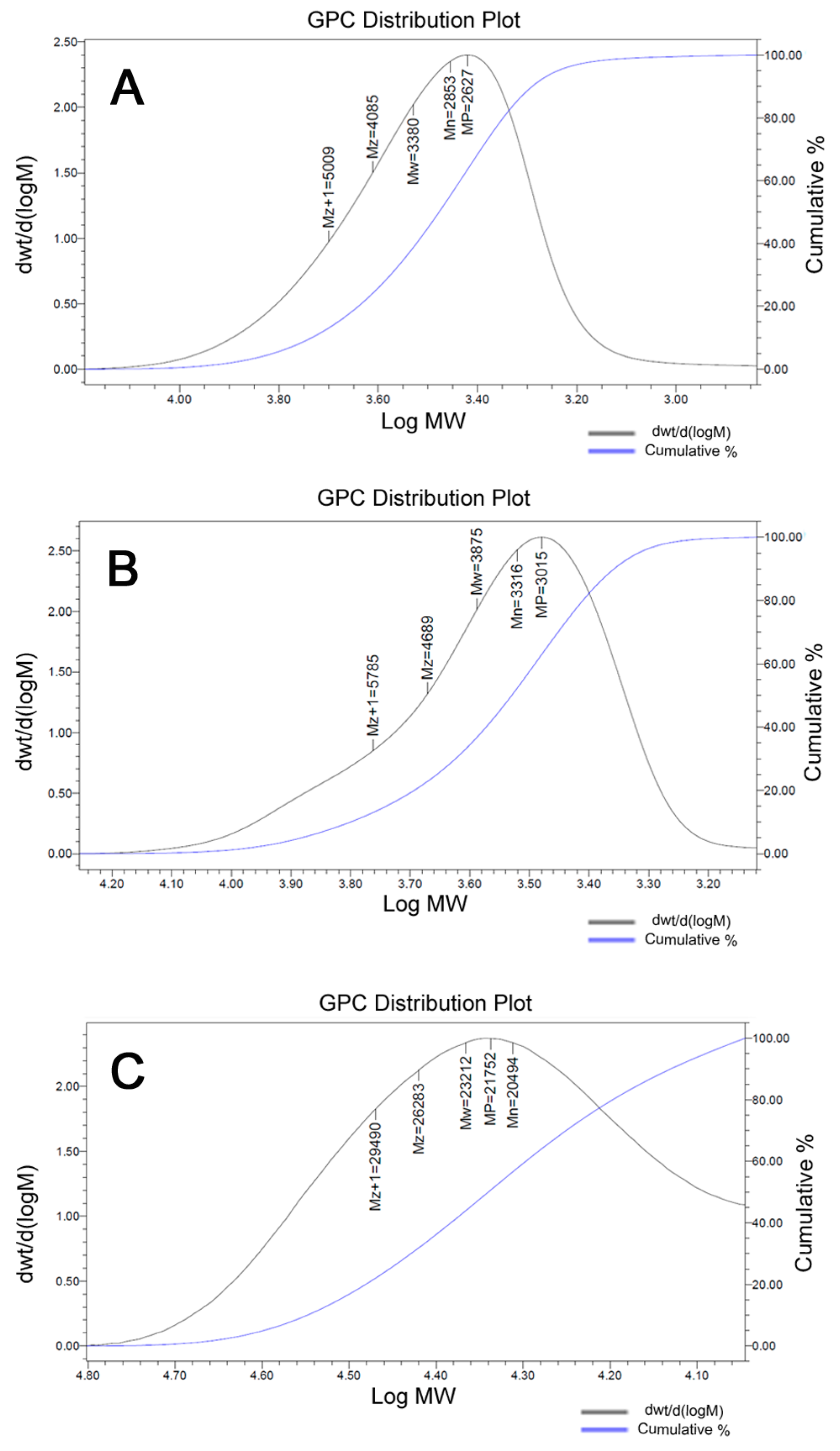
3.2. Synthesis of HEMA-PLA and HEMA-PLA-Cisplatin Conjugate
3.3. Synthesis of Poly (Mal-HPLA-cPt) by RAFT Polymerization
3.4. Synthesis of PTX-Loaded Mal-PEG-HPLA-cPt Nanoparticles (PTX-PHPLA-cPt NPs) and CTX-Attached PTX-PHPLA-cPt NPs
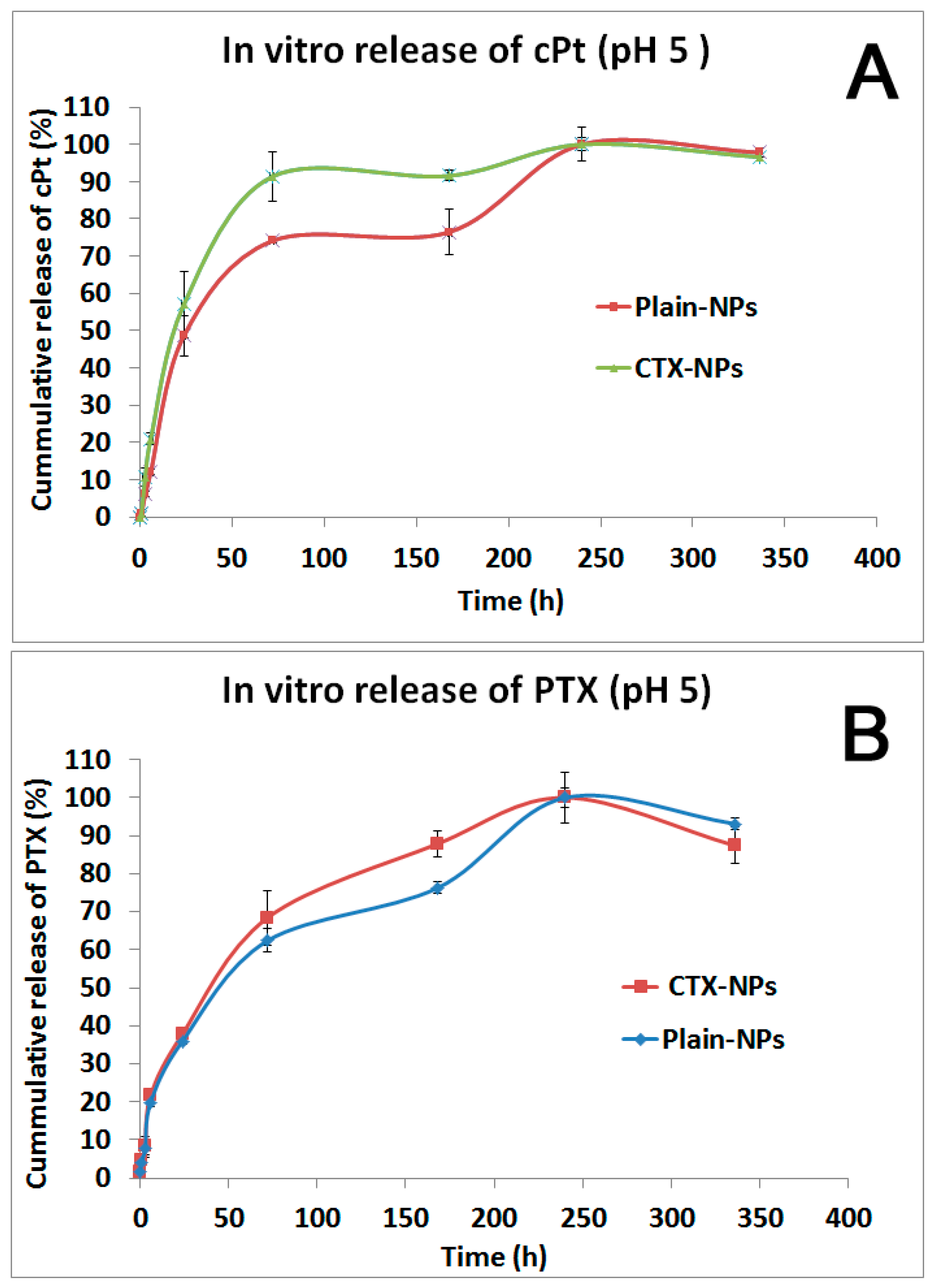
3.5. In Vitro Drug Release Study
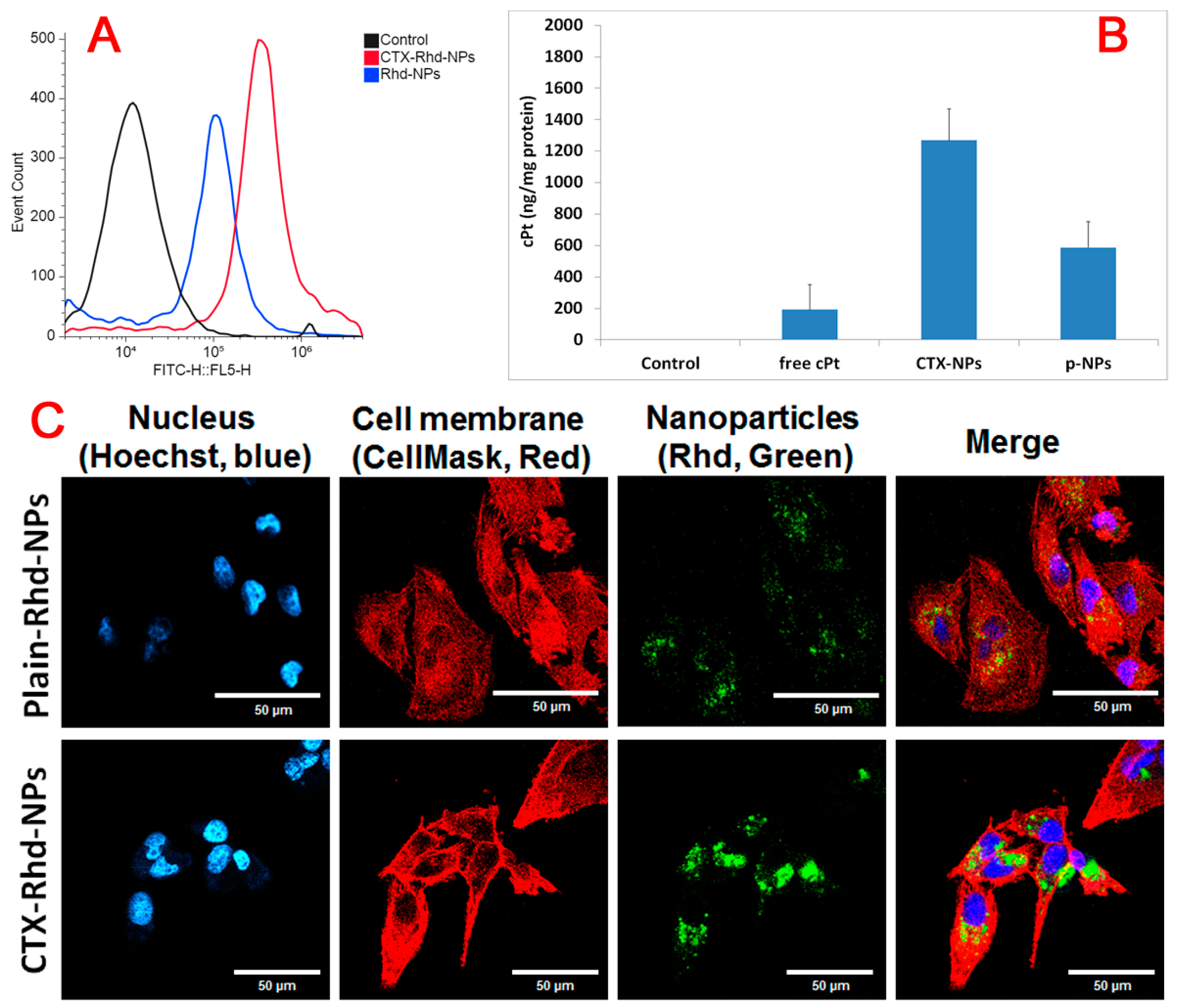
3.6. Cellular Uptake Study by Flow Cytometry and AAS
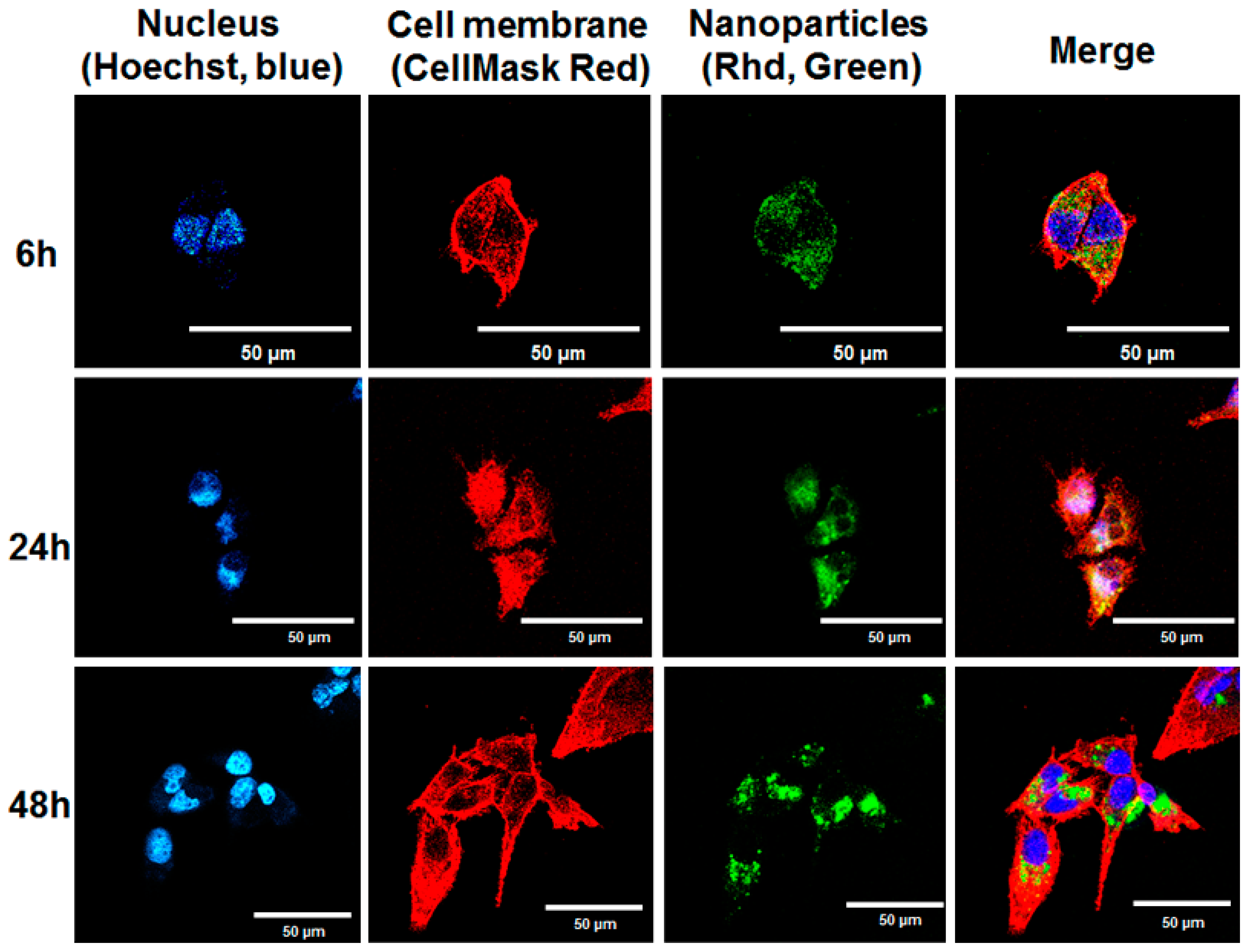
3.7. Confocal Microscope Study
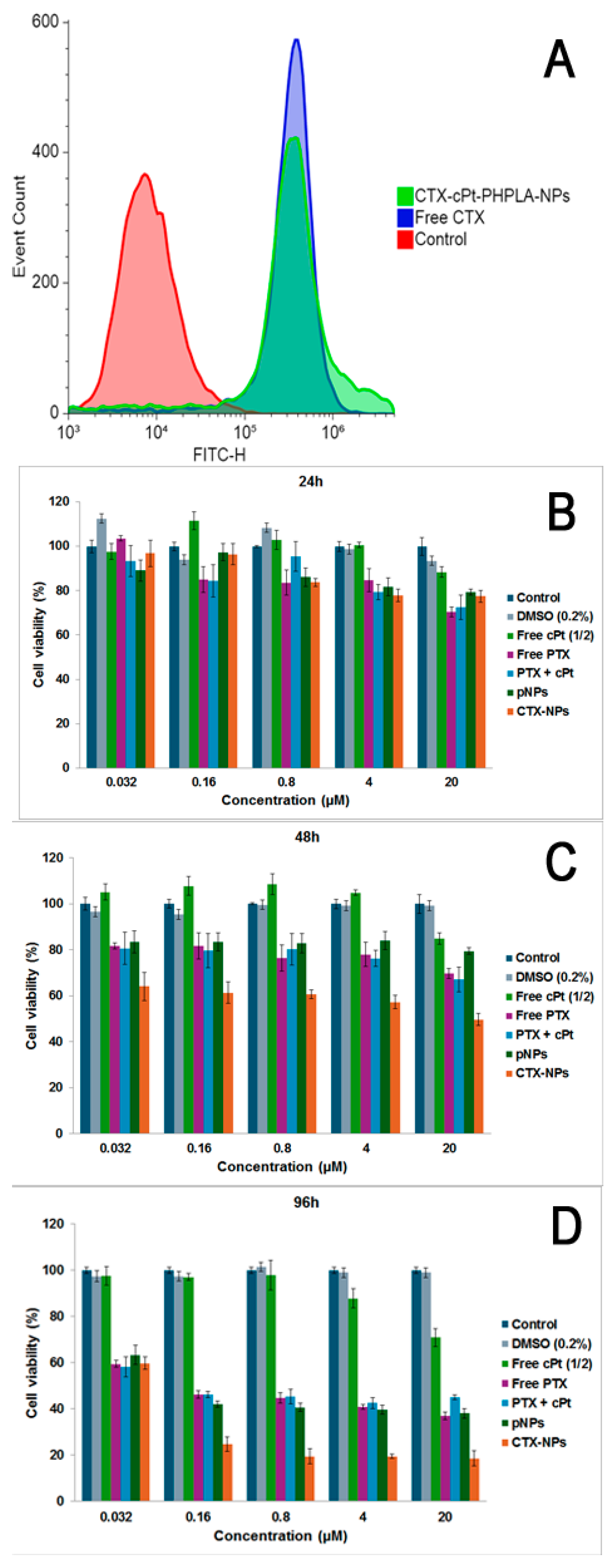
3.8. Cell Binding of CTX-NPs and Cytotoxicity on MDA-MB-231 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Battogtokh, G.; Obidiro, O.; Akala, E.O. Recent Developments in Combination Immunotherapy with Other Therapies and Nanoparticle-Based Therapy for Triple-Negative Breast Cancer (TNBC). Cancers 2024, 16, 2012. [Google Scholar] [CrossRef] [PubMed]
- Howard, F.M.; Olopade, O.I. Epidemiology of Triple-Negative Breast Cancer: A Review. Cancer J. 2021, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, K. Theranostics for Triple-Negative Breast Cancer. Diagnostics 2023, 13, 272. [Google Scholar] [CrossRef]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar]
- Medina, M.A.; Oza, G.; Sharma, A.; Arriaga, L.G.; Hernandez Hernandez, J.M.; Rotello, V.M.; Ramirez, J.T. Triple-Negative Breast Cancer: A Review of Conventional and Advanced Therapeutic Strategies. Int. J. Environ. Res. Public Health 2020, 17, 2078. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar]
- von Minckwitz, G.; Schneeweiss, A.; Loibl, S.; Salat, C.; Denkert, C.; Rezai, M.; Blohmer, J.U.; Jackisch, C.; Paepke, S.; Gerber, B.; et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 2014, 15, 747–756. [Google Scholar]
- Sikov, W.M.; Berry, D.A.; Perou, C.M.; Singh, B.; Cirrincione, C.T.; Tolaney, S.M.; Kuzma, C.S.; Pluard, T.J.; Somlo, G.; Port, E.R.; et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J Clin Oncol. 2015, 33, 13–21. [Google Scholar] [PubMed]
- Hu, X.C.; Zhang, J.; Xu, B.H.; Cai, L.; Ragaz, J.; Wang, Z.H.; Wang, B.Y.; Teng, Y.E.; Tong, Z.S.; Pan, Y.Y.; et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Inoue, K.; Kaneko, K.; Ito, Y.; Tsugawa, K.; Hasegawa, A.; Nakagawa, S.; Kuratomi, H.; Tamura, K. Subgroup analysis of Japanese patients in a Phase 3 study of atezolizumab in advanced triple-negative breast cancer (IMpassion130). Jpn. J. Clin. Oncol. 2019, 49, 1083–1091. [Google Scholar] [CrossRef]
- Foldi, J.; Kahn, A.; Silber, A.; Qing, T.; Reisenbichler, E.; Fischbach, N.; Persico, J.; Adelson, K.; Katoch, A.; Chagpar, A.; et al. Clinical Outcomes and Immune Markers by Race in a Phase I/II Clinical Trial of Durvalumab Concomitant with Neoadjuvant Chemotherapy in Early-Stage TNBC. Clin. Cancer Res. 2022, 28, 3720–3728. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Chan, A.; Dirix, L.; O’Shaughnessy, J.; Hegg, R.; Manikhas, A.; Shtivelband, M.; Krivorotko, P.; Batista Lopez, N.; Campone, M.; et al. A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2- advanced breast cancer (BELLE-4). Ann. Oncol. 2017, 28, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Dent, R.; Im, S.A.; Espie, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Anand, K.; Patel, T.; Niravath, P.; Rodriguez, A.; Darcourt, J.; Belcheva, A.; Boone, T.; Ensor, J.; Chang, J. Targeting mTOR and DNA repair pathways in residual triple negative breast cancer post neoadjuvant chemotherapy. Sci. Rep. 2021, 11, 82. [Google Scholar] [CrossRef]
- Rugo, H.S.; Olopade, O.I.; DeMichele, A.; Yau, C.; van’t Veer, L.J.; Buxton, M.B.; Hogarth, M.; Hylton, N.M.; Paoloni, M.; Perlmutter, J.; et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N. Engl. J. Med. 2016, 375, 23–34. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Huang, L.; Liu, Q.; Chen, S.; Shao, Z.M. Cisplatin versus carboplatin in combination with paclitaxel as neoadjuvant regimen for triple negative breast cancer. Oncotargets Ther. 2017, 10, 5739–5744. [Google Scholar] [CrossRef]
- Tian, H.; Ma, D.; Tan, X.; Yan, W.; Wu, X.; He, C.; Zhong, L.; Zhang, Y.; Yu, B.; Zhang, Y.; et al. Platinum and Taxane Based Adjuvant and Neoadjuvant Chemotherapy in Early Triple-Negative Breast Cancer: A Narrative Review. Front. Pharmacol. 2021, 12, 770663. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Li, Z.; Shen, G.; Wang, T.; Li, J.; Wang, M.; Liu, Z.; Zhao, F.; Ren, D.; Zhao, J. Efficacy and safety of first-line treatment for metastatic triple-negative breast cancer: A network meta-analysis. Cancer Pathog. Ther. 2024, 2, 81–90. [Google Scholar] [PubMed]
- Petrelli, F.; Barni, S.; Bregni, G.; de Braud, F.; Di Cosimo, S. Platinum salts in advanced breast cancer: A systematic review and meta-analysis of randomized clinical trials. Breast Cancer Res. Treat. 2016, 160, 425–437. [Google Scholar]
- Aslan, B.; Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Nanotechnology in cancer therapy. J. Drug Target 2013, 21, 904–913. [Google Scholar]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar]
- Sung, Y.K.; Kim, S.W. Recent advances in polymeric drug delivery systems. Biomater. Res. 2020, 24, 12. [Google Scholar]
- Kim, J.; Pramanick, S.; Lee, D.; Park, H.; Kim, W.J. Polymeric biomaterials for the delivery of platinum-based anticancer drugs. Biomater. Sci. 2015, 3, 1002–1017. [Google Scholar]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [PubMed]
- Baselga, J.; Gomez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pego, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Milane, L.; Duan, Z.; Amiji, M. Therapeutic efficacy and safety of paclitaxel/lonidamine loaded EGFR-targeted nanoparticles for the treatment of multi-drug resistant cancer. PLoS ONE 2011, 6, e24075. [Google Scholar]
- Kutty, R.V.; Feng, S.S. Cetuximab conjugated vitamin E TPGS micelles for targeted delivery of docetaxel for treatment of triple negative breast cancers. Biomaterials 2013, 34, 10160–10171. [Google Scholar]
- Xiao, H.H.; Qi, R.G.; Liu, S.; Hu, X.L.; Duan, T.C.; Zheng, Y.H.; Huang, Y.B.; Jing, X.B. Biodegradable polymer—Cisplatin(IV) conjugate as a pro-drug of cisplatin(II). Biomaterials 2011, 32, 7732–7739. [Google Scholar]
- Shi, Y.; Liu, S.A.; Kerwood, D.J.; Goodisman, J.; Dabrowiak, J.C. Pt(IV) complexes as prodrugs for cisplatin. J. Inorg. Biochem. 2012, 107, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Gatti, S.; Agostini, A.; Ferrari, R.; Moscatelli, D. Synthesis and Nanoprecipitation of HEMA-CLn Based Polymers for the Production of Biodegradable Nanoparticles. Polymers 2017, 9, 389. [Google Scholar] [CrossRef]
- Battogtokh, G.; Liu, H.B.; Bae, S.M.; Chaturvedi, P.K.; Kim, Y.W.; Kim, I.W.; Ahn, W.S. In vitro phototoxicity and dark-toxicity of a novel synthesized pyropheophorbide-a-paclitaxel conjugate against cancer cell lines. J. Porphyr. Phthalocyanines 2012, 16, 1006–1014. [Google Scholar]
- Colombo, C.; Gatti, S.; Ferrari, R.; Casalini, T.; Cuccato, D.; Morosi, L.; Zucchetti, M.; Moscatelli, D. Self-assembling amphiphilic PEGylated block copolymers obtained through RAFT polymerization for drug-delivery applications. J. Appl. Polym. Sci. 2016, 133, 43084. [Google Scholar]
- Duan, Z.; Zhang, Y.; Zhu, H.; Sun, L.; Cai, H.; Li, B.; Gong, Q.; Gu, Z.; Luo, K. Stimuli-Sensitive Biodegradable and Amphiphilic Block Copolymer-Gemcitabine Conjugates Self-Assemble into a Nanoscale Vehicle for Cancer Therapy. ACS Appl. Mater. Interfaces 2017, 9, 3474–3486. [Google Scholar]
- Bahmani, B.; Uehara, M.; Ordikhani, F.; Li, X.; Jiang, L.; Banouni, N.; Ichimura, T.; Kasinath, V.; Eskandari, S.K.; Annabi, N.; et al. Ectopic high endothelial venules in pancreatic ductal adenocarcinoma: A unique site for targeted delivery. EBioMedicine 2018, 38, 79–88. [Google Scholar]
- Chen, P.M.; Katsuyama, E.; Satyam, A.; Li, H.; Rubio, J.; Jung, S.; Andrzejewski, S.; Becherer, J.D.; Tsokos, M.G.; Abdi, R.; et al. CD38 reduces mitochondrial fitness and cytotoxic T cell response against viral infection in lupus patients by suppressing mitophagy. Sci. Adv. 2022, 8, eabo4271. [Google Scholar]
- Davydova, M.; Dewaele Le Roi, G.; Adumeau, P.; Zeglis, B.M. Synthesis and Bioconjugation of Thiol-Reactive Reagents for the Creation of Site-Selectively Modified Immunoconjugates. J. Vis. Exp. 2019, 145, 10-3791. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.M.; Sun, Y.; Chen, H.L.; Zhou, L.Y. Cetuximab-decorated and NIR-activated Nanoparticles Based on Platinum(IV)-prodrug: Preparation, Characterization and In-vitro Anticancer Activity in Epidermoid Carcinoma Cells. Iran. J. Pharm. Res. 2021, 20, 371–383. [Google Scholar]
- Yang, J.; Lee, C.H.; Park, J.; Seo, S.; Lim, E.K.; Song, Y.J.; Suh, J.S.; Yoon, H.G.; Huh, Y.M.; Haam, S. Antibody conjugated magnetic PLGA nanoparticles for diagnosis and treatment of breast cancer. J. Mater. Chem. 2007, 17, 2695–2699. [Google Scholar]
- Berko, Y.A.; Funmilola, A.F.; Akala, E.O. Fabrication of Paclitaxel and 17AAG-loaded Poly-epsilon-Caprolactone Nanoparticles for Breast Cancer Treatment. J. Pharm. Drug Deliv. Res. 2021, 10, 196. [Google Scholar]
- Battogtokh, G.; Joo, Y.; Abuzar, S.M.; Park, H.; Hwang, S.J. Gelatin Coating for the Improvement of Stability and Cell Uptake of Hydrophobic Drug-Containing Liposomes. Molecules 2022, 27, 1041. [Google Scholar] [CrossRef] [PubMed]
- Battogtokh, G.; Ko, Y.T. Self-assembled chitosan-ceramide nanoparticle for enhanced oral delivery of paclitaxel. Pharm. Res. 2014, 31, 3019–3030. [Google Scholar]
- Raghavan, R.; Mulligan, J.A. Low-level (PPB) determination of cisplatin in cleaning validation (rinse water) samples. I. An atomic absorption spectrophotometric method. Drug Dev. Ind. Pharm. 2000, 26, 423–428. [Google Scholar]
- Adesina, S.K.; Holly, A.; Kramer-Marek, G.; Capala, J.; Akala, E.O. Polylactide-based paclitaxel-loaded nanoparticles fabricated by dispersion polymerization: Characterization, evaluation in cancer cell lines, and preliminary biodistribution studies. J. Pharm. Sci. 2014, 103, 2546–2555. [Google Scholar]
- Battogtokh, G.; Kang, J.H.; Ko, Y.T. Long-circulating self-assembled cholesteryl albumin nanoparticles enhance tumor accumulation of hydrophobic anticancer drug. Eur. J. Pharm. Biopharm. 2015, 96, 96–105. [Google Scholar] [PubMed]
- Kolishetti, N.; Dhar, S.; Valencia, P.M.; Lin, L.Q.; Karnik, R.; Lippard, S.J.; Langer, R.; Farokhzad, O.C. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 17939–17944. [Google Scholar]
- Cao, Z.; Li, W.; Liu, R.; Li, X.; Li, H.; Liu, L.; Chen, Y.; Lv, C.; Liu, Y. pH- and enzyme-triggered drug release as an important process in the design of anti-tumor drug delivery systems. Biomed. Pharmacother. 2019, 118, 109340. [Google Scholar]
- Zhou, Y.; Li, N.; Qiu, Z.; Lu, X.; Fang, M.; Chen, X.; Ren, L.; Wang, G.; Ouyang, P. Superior anti-neoplastic activities of triacontanol-PEG conjugate: Synthesis, characterization and biological evaluations. Drug Deliv. 2018, 25, 1546–1559. [Google Scholar] [PubMed]
- Semsarilar, M.; Abetz, V. Polymerizations by RAFT: Developments of the Technique and Its Application in the Synthesis of Tailored (Co)polymers. Macromol. Chem. Phys. 2021, 222, 2000311. [Google Scholar]
- Desai, N.; Rana, D.; Salave, S.; Benival, D.; Khunt, D.; Prajapati, B.G. Achieving Endo/Lysosomal Escape Using Smart Nanosystems for Efficient Cellular Delivery. Molecules 2024, 29, 3131. [Google Scholar] [CrossRef]
- Anwar, M.; Muhammad, F.; Akhtar, B. Biodegradable nanoparticles as drug delivery devices. J. Drug Deliv. Sci. Technol. 2021, 64, 102638. [Google Scholar]
- Panyam, J.; Zhou, W.Z.; Prabha, S.; Sahoo, S.K.; Labhasetwar, V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002, 16, 1217–1226. [Google Scholar]
- Wang, Y.; Sui, Y.; Tao, Y. Gambogic acid increases the sensitivity to paclitaxel in drug-resistant triple-negative breast cancer via the SHH signaling pathway. Mol. Med. Rep. 2019, 20, 4515–4522. [Google Scholar]
- Pauzi, A.Z.; Yeap, S.K.; Abu, N.; Lim, K.L.; Omar, A.R.; Aziz, S.A.; Chow, A.L.; Subramani, T.; Tan, S.G.; Alitheen, N.B. Combination of cisplatin and bromelain exerts synergistic cytotoxic effects against breast cancer cell line MDA-MB-231 in vitro. Chin. Med. 2016, 11, 46. [Google Scholar] [CrossRef]
- Haghnavaz, N.; Asghari, F.; Elieh Ali Komi, D.; Shanehbandi, D.; Baradaran, B.; Kazemi, T. HER2 positivity may confer resistance to therapy with paclitaxel in breast cancer cell lines. Artif. Cells Nanomed. Biotechnol. 2018, 46, 518–523. [Google Scholar] [PubMed]
- Han, Y.; Yu, X.; Li, S.; Tian, Y.; Liu, C. New Perspectives for Resistance to PARP Inhibitors in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 578095. [Google Scholar]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 240–249. [Google Scholar]
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021, 70, 607–617. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticle | Mean Diameter (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) |
|---|---|---|---|
| Plain PTX-PHPLA-cPt-NPs (p-NPs) | 161.6 ± 3.2 | 0.206 ± 0.007 | −12.41 ± 0.37 |
| CTX-PTX-PHPLA-cPt-NPs (CTX-NPs) | 198.7 ± 1.3 | 0.047 ± 0.004 | −41.32 ± 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battogtokh, G.; Akala, E.O. Development of Multifunctional Targeted Dual-Loaded Polymeric Nanoparticles for Triple-Negative Breast Cancer Treatment. Pharmaceutics 2025, 17, 425. https://doi.org/10.3390/pharmaceutics17040425
Battogtokh G, Akala EO. Development of Multifunctional Targeted Dual-Loaded Polymeric Nanoparticles for Triple-Negative Breast Cancer Treatment. Pharmaceutics. 2025; 17(4):425. https://doi.org/10.3390/pharmaceutics17040425
Chicago/Turabian StyleBattogtokh, Gantumur, and Emmanuel O. Akala. 2025. "Development of Multifunctional Targeted Dual-Loaded Polymeric Nanoparticles for Triple-Negative Breast Cancer Treatment" Pharmaceutics 17, no. 4: 425. https://doi.org/10.3390/pharmaceutics17040425
APA StyleBattogtokh, G., & Akala, E. O. (2025). Development of Multifunctional Targeted Dual-Loaded Polymeric Nanoparticles for Triple-Negative Breast Cancer Treatment. Pharmaceutics, 17(4), 425. https://doi.org/10.3390/pharmaceutics17040425