
A Sequential Ultrafiltration Method to Enhance the Accuracy and Throughput in Plasma Protein Binding Tests

 , ,
, ,
Abstract
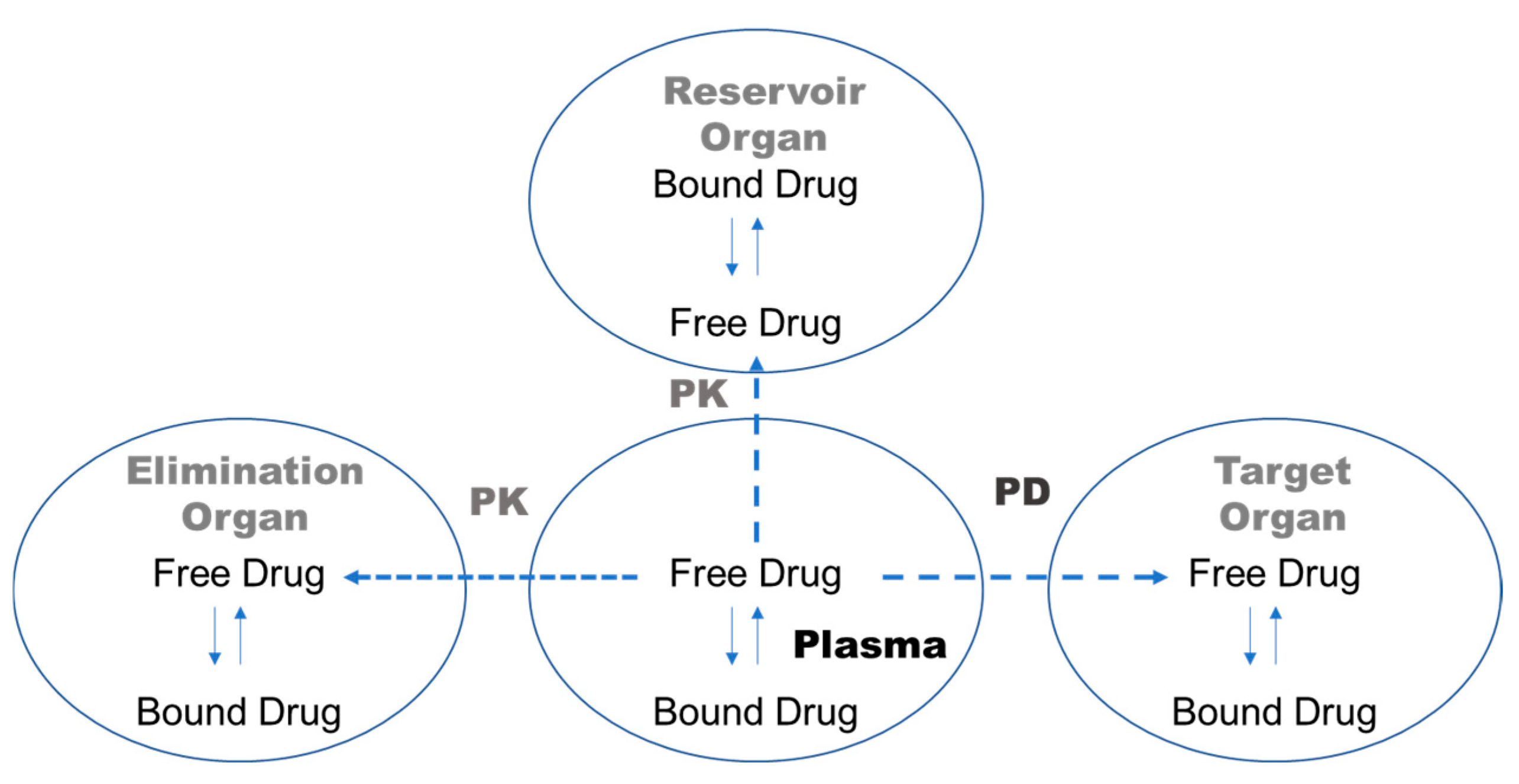
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Plasma Sample Preparation
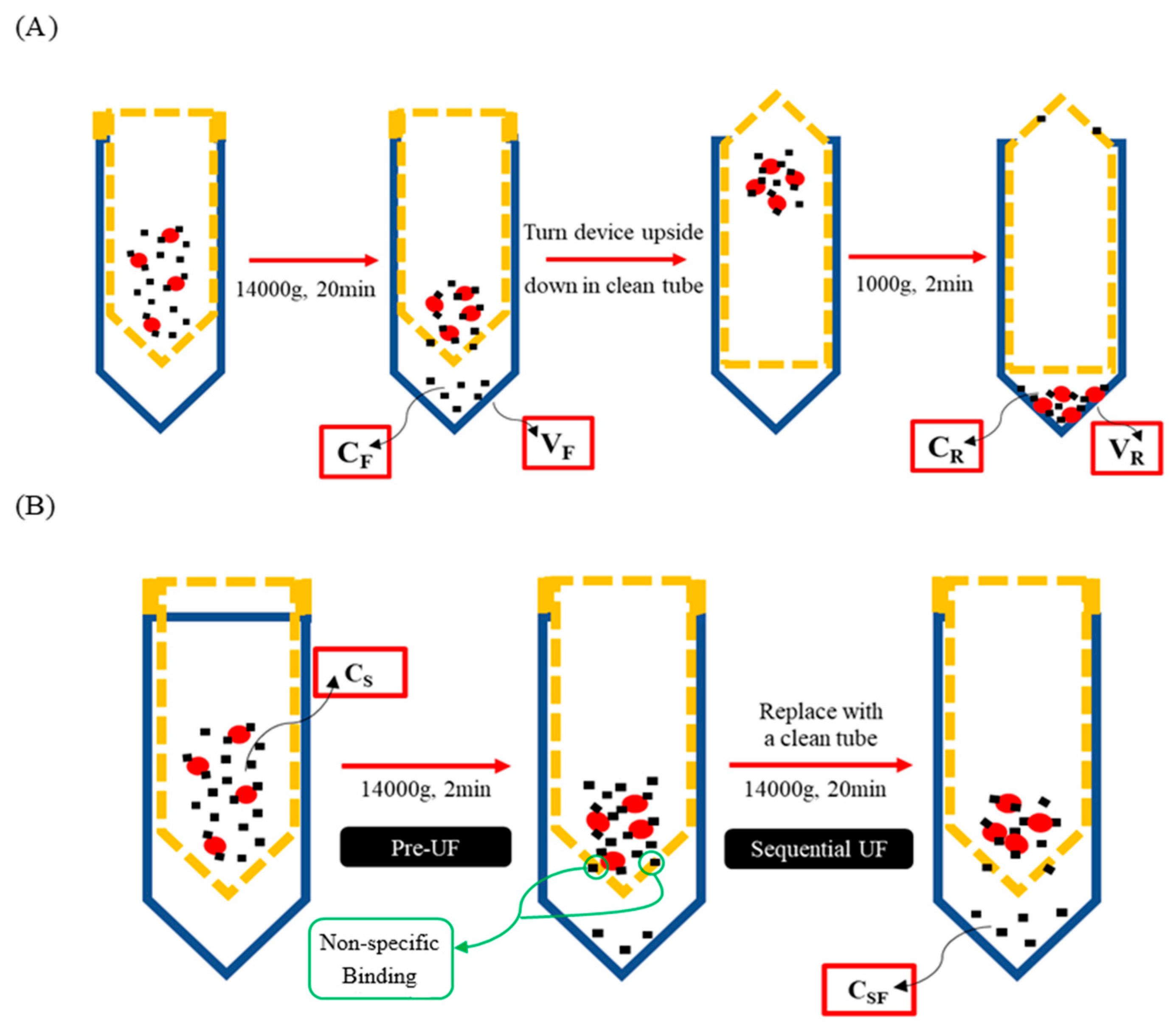
2.3. Conventional UF with and Without NSB Correction
2.4. Mass Balance UF
2.5. Sequential UF
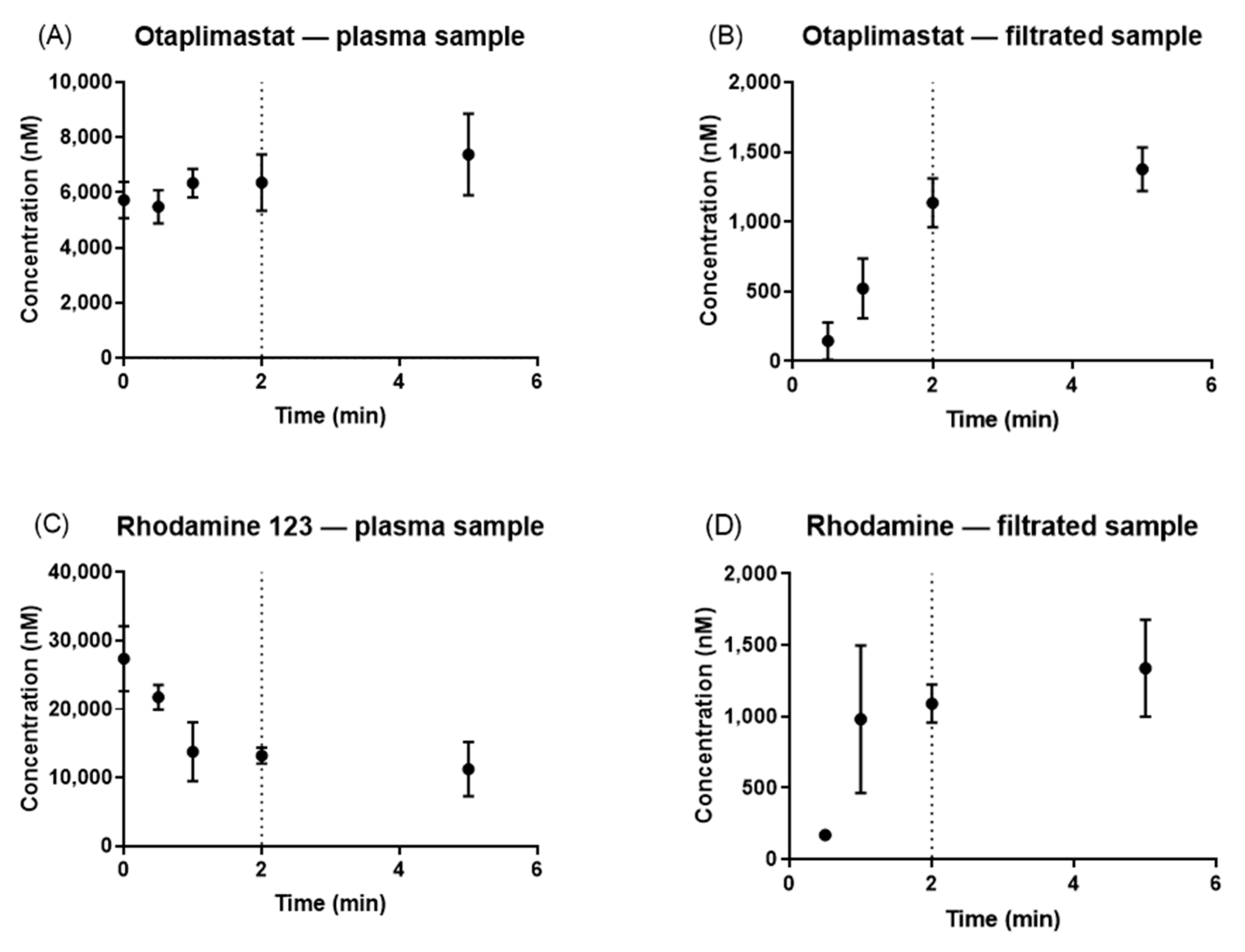
2.6. Determination of Optimal Pre-UF Time for Sequential UF
2.7. Quantitative Analysis
2.8. Comparison of Methods
2.9. Statistical Analysis
3. Results
3.1. Optimal Pre-UF Time
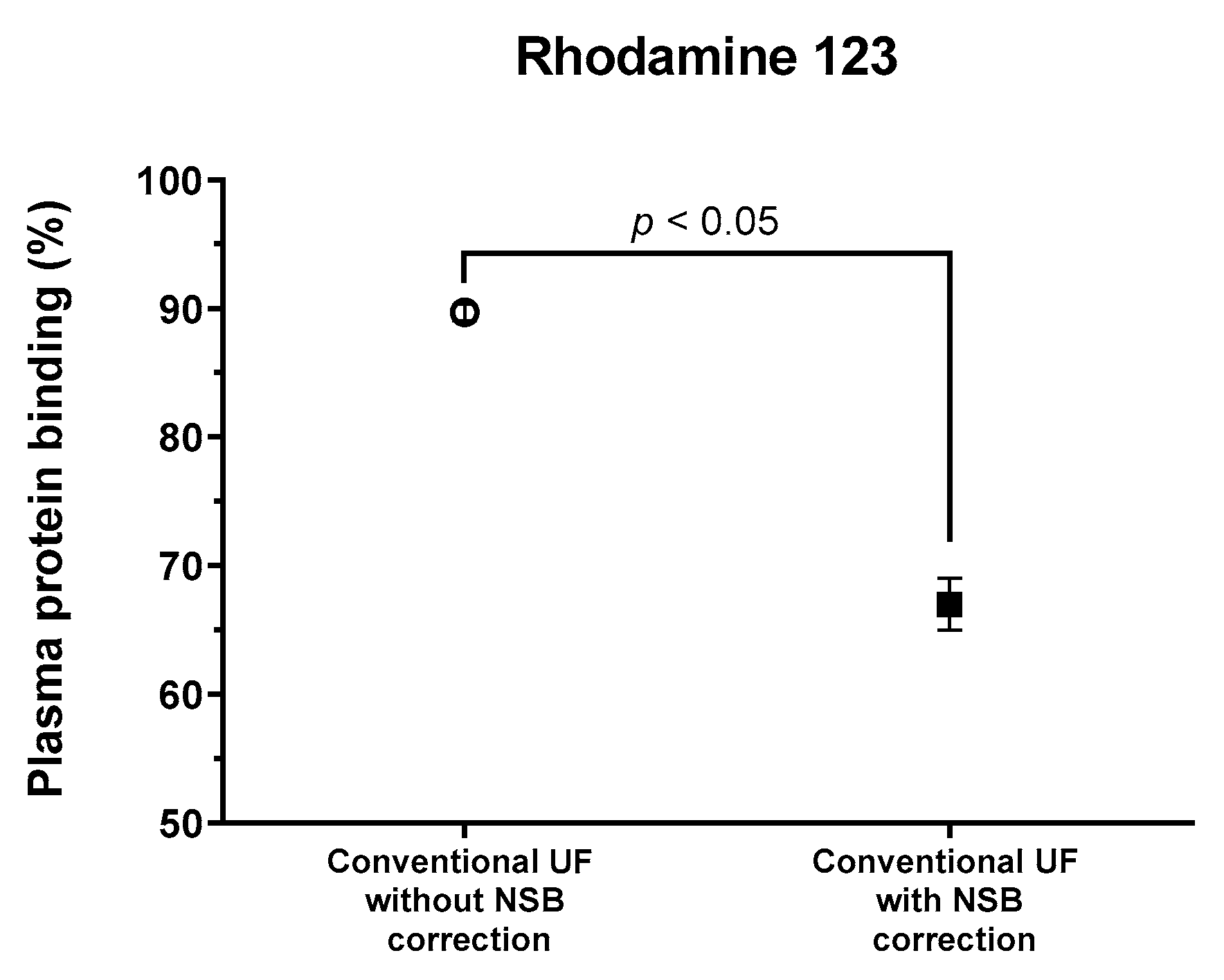
3.2. Effects of NSB on the PPB of Rhodamine 123
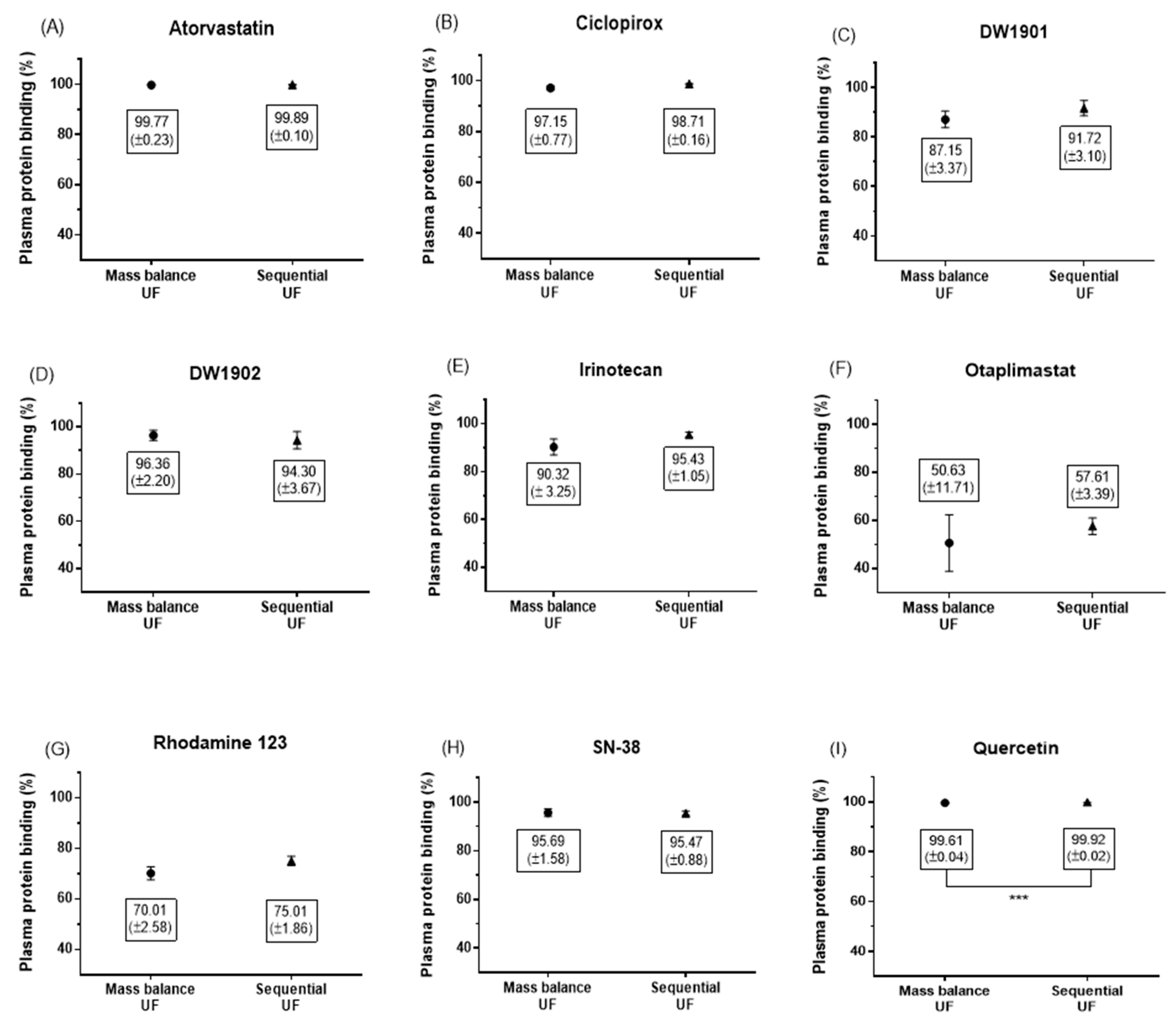
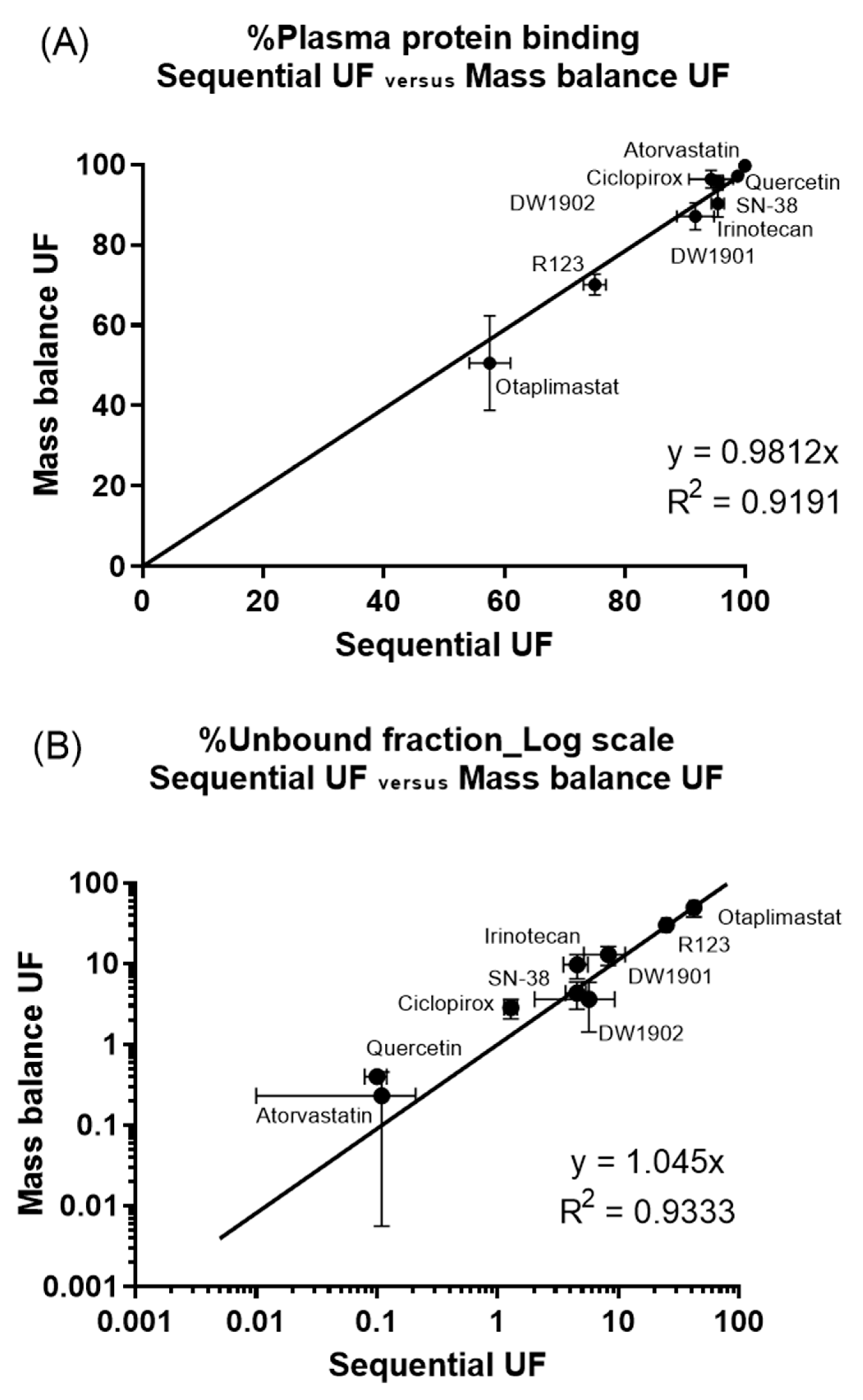
3.3. Comparison of Mass Balance UF and Sequential UF
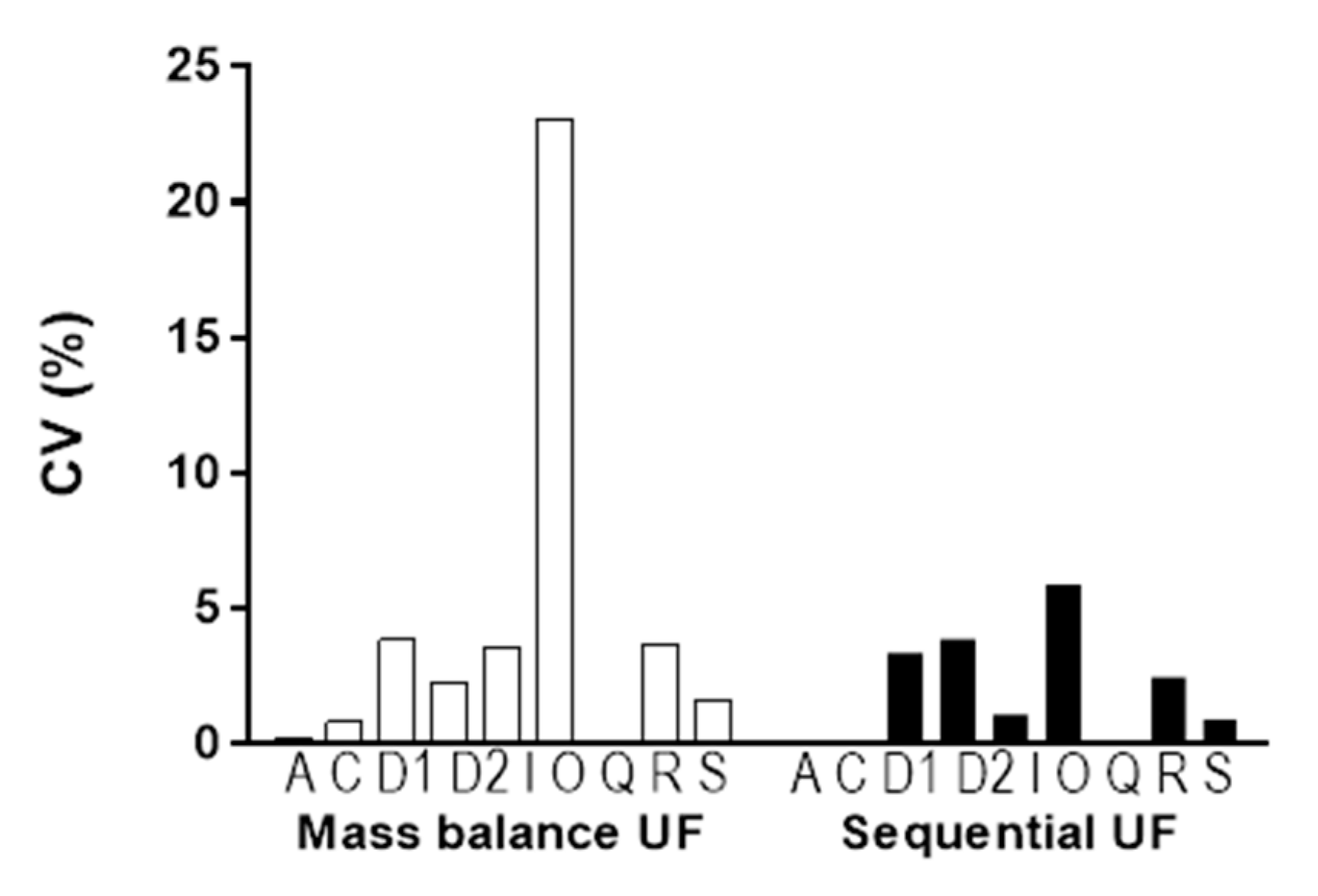
3.4. Comparison of Variance in Results
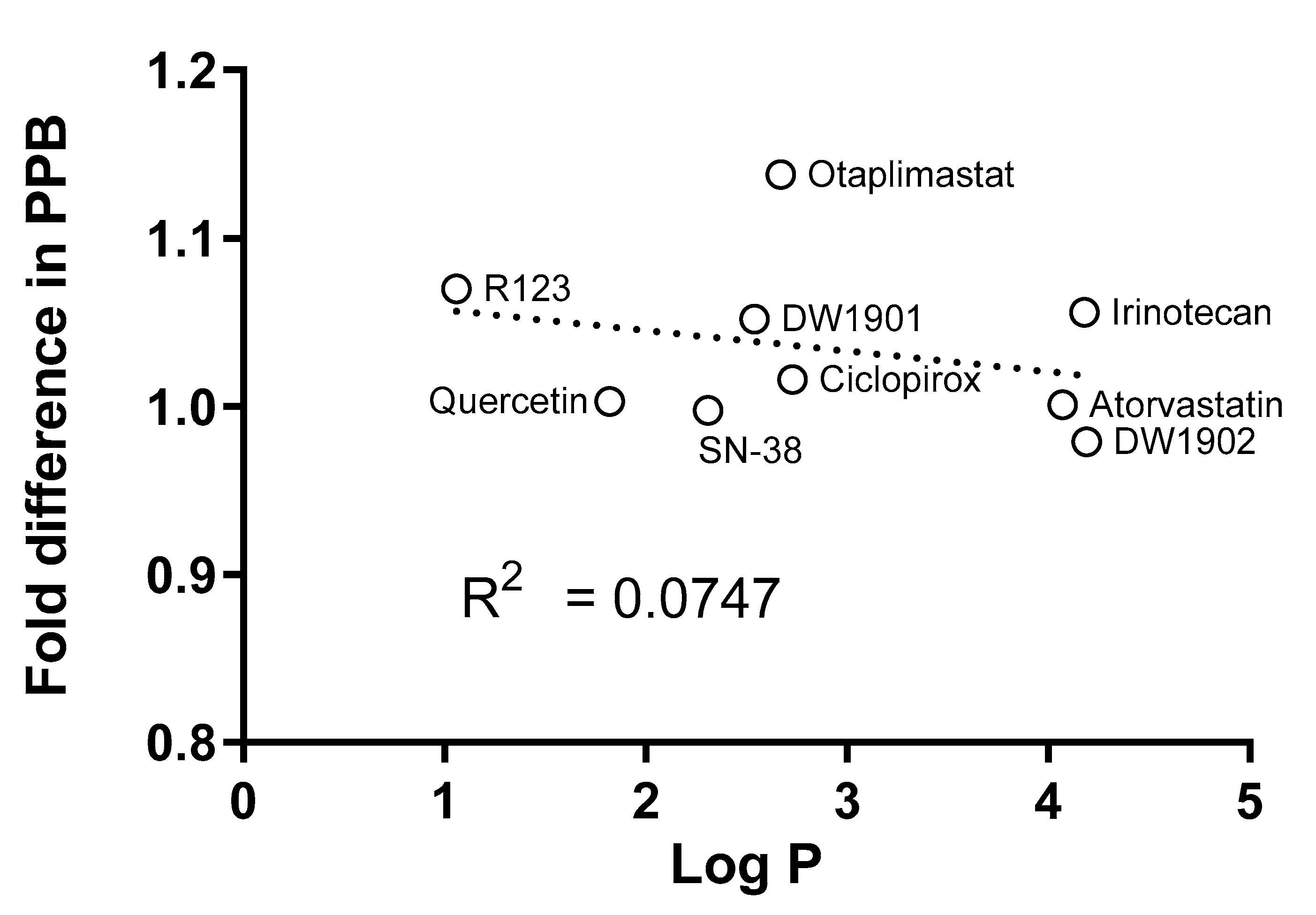
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Di, L. An update on the importance of plasma protein binding in drug discovery and development. Expert. Opin. Drug Discov. 2021, 16, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Summerfield, S.G.; Yates, J.W.T.; Fairman, D.A. Free Drug Theory—No Longer Just a Hypothesis? Pharm. Res. 2022, 39, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.A.; Stresser, D.M.; Kalvass, J.C. Plasma protein-mediated uptake and contradictions to the free drug hypothesis: A critical review. Drug Metab. Rev. 2023, 55, 205–238. [Google Scholar] [CrossRef] [PubMed]
- Francis, L.J.; Houston, J.B.; Hallifax, D. Impact of Plasma Protein Binding in Drug Clearance Prediction: A Data Base Analysis of Published Studies and Implications for In Vitro-In Vivo Extrapolation. Drug Metab. Dispos. 2021, 49, 188–201. [Google Scholar] [CrossRef]
- Deb, P.K.; Al-Attraqchi, O.; Prasad, M.R.; Tekade, R.K. Chapter 11—Protein and Tissue Binding: Implication on Pharmacokinetic Parameters. In Dosage Form Design Considerations; Tekade, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 371–399. [Google Scholar] [CrossRef]
- Dimitrijevic, D.; Fabian, E.; Funk-Weyer, D.; Landsiedel, R. Rapid equilibrium dialysis, ultrafiltration or ultracentrifugation? Evaluation of methods to quantify the unbound fraction of substances in plasma. Biochem. Biophys. Res. Commun. 2023, 651, 114–120. [Google Scholar] [CrossRef]
- Toma, C.-M.; Imre, S.; Vari, C.-E.; Muntean, D.-L.; Tero-Vescan, A. Ultrafiltration Method for Plasma Protein Binding Studies and Its Limitations. Processes 2021, 9, 382. [Google Scholar] [CrossRef]
- Seyfinejad, B.; Ozkan, S.A.; Jouyban, A. Recent advances in the determination of unbound concentration and plasma protein binding of drugs: Analytical methods. Talanta 2021, 225, 122052. [Google Scholar] [CrossRef]
- Boer, T.d.; Meijering, H. Equilibrium Dialysis, Ultracentrifugation, and Ultrafiltration in LC-MS Bioanalysis. In Sample Preparation in LC-MS Bioanalysis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2019; pp. 45–51. [Google Scholar] [CrossRef]
- Illamola, S.M.; Labat, L.; Benaboud, S.; Tubiana, R.; Warszawski, J.; Treluyer, J.M.; Hirt, D. Determination of total and unbound concentrations of lopinavir in plasma using liquid chromatography-tandem mass spectrometry and ultrafiltration methods. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2014, 965, 216–223. [Google Scholar] [CrossRef]
- Lee, K.J.; Mower, R.; Hollenbeck, T.; Castelo, J.; Johnson, N.; Gordon, P.; Sinko, P.J.; Holme, K.; Lee, Y.H. Modulation of nonspecific binding in ultrafiltration protein binding studies. Pharm. Res. 2003, 20, 1015–1021. [Google Scholar] [CrossRef]
- Ratnaningsih, E.; Reynard, R.; Khoiruddin, K.; Wenten, I.G.; Boopathy, R. Recent Advancements of UF-Based Separation for Selective Enrichment of Proteins and Bioactive Peptides—A Review. Appl. Sci. 2021, 11, 1078. [Google Scholar] [CrossRef]
- Taylor, S.; Harker, A. Modification of the ultrafiltration technique to overcome solubility and non-specific binding challenges associated with the measurement of plasma protein binding of corticosteroids. J. Pharm. Biomed. Anal. 2006, 41, 299–303. [Google Scholar] [CrossRef]
- Wang, C.; Williams, N.S. A mass balance approach for calculation of recovery and binding enables the use of ultrafiltration as a rapid method for measurement of plasma protein binding for even highly lipophilic compounds. J. Pharm. Biomed. Anal. 2013, 75, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Toma, C.-M.; Farczádi, L.; Ion, V.; Muntean, D.-L.; Imre, S. Comparison of Different Ultrafiltration Devices for the Study of Plasma Protein Binding of Carvedilol. Acta Biol. Marisiensis 2023, 6, 1–10. [Google Scholar] [CrossRef]
- Kratzer, A.; Liebchen, U.; Schleibinger, M.; Kees, M.G.; Kees, F. Determination of free vancomycin, ceftriaxone, cefazolin and ertapenem in plasma by ultrafiltration: Impact of experimental conditions. J. Chromatography. B Anal. Technol. Biomed. Life Sci. 2014, 961, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Vora, M.M. Chromatography of Rhodamine 123 and Rhodamine 110 on Reverse-Phase Liquid Chromatographic Column. J. Liq. Chromatogr. 1989, 12, 583–593. [Google Scholar] [CrossRef]
- Shin, E.; Shin, N.; Oh, J.-H.; Lee, Y.-J. High-Dose Metformin May Increase the Concentration of Atorvastatin in the Liver by Inhibition of Multidrug Resistance-Associated Protein 2. J. Pharm. Sci. 2017, 106, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Escarrone, A.L.V.; Bittencourt, C.F.; Laporta, L.V.; dos Santos, M.R.; Primel, E.G.; Caldas, S.S. LC–UV Method with Pre-Column Derivatization for the Determination of Ciclopirox Olamine in Raw Material and Topical Solution. Chromatographia 2008, 67, 967–971. [Google Scholar] [CrossRef]
- Lee, Y.-J. Kyung Hee University, Seoul, Republic of Korea, 2018, manuscript in preparation.
- Zhang, W.; Dutschman, G.E.; Li, X.; Ye, M.; Cheng, Y.-C. Quantitation of Irinotecan and its two major metabolites using a liquid chromatography-electrospray ionization tandem mass spectrometric. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3038–3044. [Google Scholar] [CrossRef]
- Lee, S.; Kim, M.; Oh, J.-H.; Lee, J.H.; Shin, N.; Park, T.; Lee, J.H.; Kim, M.C.; Lee, Y.-J. Optimized liquid chromatography-tandem mass spectrometry for Otaplimastat quantification in rat plasma and brain tissue. J. Chromatogr. Sci. 2019, 57, 258–264. [Google Scholar] [CrossRef]
- D’Esposito, F.; Tattam, B.N.; Ramzan, I.; Murray, M. A liquid chromatography/electrospray ionization mass spectrometry (LC-MS/MS) assay for the determination of irinotecan (CPT-11) and its two major metabolites in human liver microsomal incubations and human plasma samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 875, 522–530. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Feng, Y.; Ouyang, H.Z.; Yu, B.; Chang, Y.X.; Pan, G.X.; Dong, G.Y.; Wang, T.; Gao, X.M. A sensitive LC-MS/MS method for simultaneous determination of six flavonoids in rat plasma: Application to a pharmacokinetic study of total flavonoids from mulberry leaves. J. Pharm. Biomed. Anal. 2013, 84, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Nishio, Y.; Ito, K.; Nakao, A.; Wang, L.; Zhao, Y.L.; Kitaichi, K.; Takagi, K.; Hasegawa, T. Effect of endotoxin on P-glycoprotein-mediated biliary and renal excretion of rhodamine-123 in rats. Antimicrob. Agents Chemother. 2001, 45, 3462–3467. [Google Scholar] [CrossRef] [PubMed]
- Urien, S.; Tillement, J.-P.; Barré, J. The Significance of Plasma-Protein Binding in Drug Research. In Pharmacokinetic Optimization in Drug Research; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001; pp. 189–197. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, L.; Grillo, J.A.; Liu, Q.; Bullock, J.M.; Moon, Y.J.; Song, P.; Brar, S.S.; Madabushi, R.; Wu, T.C.; et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 2011, 89, 259–267. [Google Scholar] [CrossRef]
- Duan, P.; Zhao, P.; Zhang, L. Physiologically Based Pharmacokinetic (PBPK) Modeling of Pitavastatin and Atorvastatin to Predict Drug-Drug Interactions (DDIs). Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 689–705. [Google Scholar] [CrossRef]
- Bu, W.; Fan, X.Q.; Sexton, H.; Heyman, I. A direct LC/MS/MS method for the determination of ciclopirox penetration across human nail plate in penetration studies. J. Pharm. Biomed. Anal. 2010, 51, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Tamura, N.; Hirano, K.; Kishino, K.; Hashimoto, K.; Amano, O.; Shimada, J.; Sakagami, H. Analysis of Type of Cell Death Induced by Topoisomerase Inhibitor SN-38 in Human Oral Squamous Cell Carcinoma Cell Lines. Anticancer. Res. 2012, 32, 4823–4832. [Google Scholar]
- Rothwell, J.A.; Day, A.J.; Morgan, M.R.A. Experimental determination of octanol-water partition coefficients of quercetin and related flavonoids. J. Agr. Food Chem. 2005, 53, 4355–4360. [Google Scholar] [CrossRef]
- Bao, X.F.; Lu, S.Y.; Liow, J.S.; Morse, C.L.; Anderson, K.B.; Zoghbi, S.S.; Innis, R.B.; Pike, V.W. [11C]Rhodamine-123: Synthesis and biodistribution in rodents. Nucl. Med. Biol. 2012, 39, 1128–1136. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | UPLC Condition | MS | Reference | |||
|---|---|---|---|---|---|---|
| Condition | ||||||
| Column | Mobile Phase | Ionization | Multiple Reaction Monitoring Transition | |||
| Mode | (m/z) | |||||
| Atorvastatin | C18 | (A) Acetonitrile | [M + H]+ | 559.7 > 440.0 | [18] | |
| (B) 0.1% formic acid in distilled water, 70:30 (v/v) | ||||||
| Ciclopirox | C18 | (A) 0.1% formic acid in 50% acetonitrile | [M + H]+ | 222.0 > 103.7 | [19] | |
| (B) Isopropanol, 90:10 (v/v) | ||||||
| DW1901 | C18 | (A) Acetonitrile | [M + H]+ | 331.0 > 141.9 | [20] | |
| (B) 0.1% formic acid in distilled water, 85:15 (v/v) | ||||||
| DW1902 | C18 | (A) Acetonitrile | [M + H]+ | 358.3 > 109.0 | [20] | |
| (B) 0.1% formic acid in distilled water, 85:15 (v/v) | ||||||
| Irinotecan | C18 | (A) 0.1% formic acid in acetonitrile | [M + H]+ | 393.3 > 349.1 | [21] | |
| (B) 0.1% formic acid in distilled water, gradient | ||||||
| Otaplimastat | C18 | (A) Acetonitrile | [M + H]+ | 535.2 > 203.0 | [22] | |
| (B) 10mM ammonium acetate, 60:40 (v/v) | ||||||
| SN-38 | C18 | (A) 0.1% formic acid in acetonitrile | [M + H]+ | 349.3 > 305.1 | [23] | |
| (B) 0.1% formic acid in distilled water, gradient | ||||||
| Quercetin | C18 | (A) 0.1% acetic acid in acetonitrile | [M − H]− | 301.0 > 151.0 | [24] | |
| (B) 1mM ammonium acetate (pH 2.5 with acetic acid), 70:30 (v/v) | ||||||
| Compound | Log p | Reference |
|---|---|---|
| Atorvastatin | 4.07 | [28] |
| Ciclopirox | 2.73 | [29] |
| DW1901 | 2.54 | a |
| DW1902 | 4.19 | a |
| Irinotecan | 4.18 | [30] |
| Otaplimastat | 2.67 | b |
| Quercetin | 1.82 | [31] |
| Rhodamine 123 | 1.06 | [32] |
| SN-38 | 2.31 | [30] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, S.H.; Kim, M.C.; Lee, H.; Oh, J.-H.; Kim, H.S.; Lee, H.; Park, T.; Lee, Y.-J. A Sequential Ultrafiltration Method to Enhance the Accuracy and Throughput in Plasma Protein Binding Tests. Pharmaceutics 2025, 17, 273. https://doi.org/10.3390/pharmaceutics17020273
Jeon SH, Kim MC, Lee H, Oh J-H, Kim HS, Lee H, Park T, Lee Y-J. A Sequential Ultrafiltration Method to Enhance the Accuracy and Throughput in Plasma Protein Binding Tests. Pharmaceutics. 2025; 17(2):273. https://doi.org/10.3390/pharmaceutics17020273
Chicago/Turabian StyleJeon, Sang Ho, Min Chang Kim, Haejun Lee, Ju-Hee Oh, Hyun Seo Kim, Heawon Lee, Taehoon Park, and Young-Joo Lee. 2025. "A Sequential Ultrafiltration Method to Enhance the Accuracy and Throughput in Plasma Protein Binding Tests" Pharmaceutics 17, no. 2: 273. https://doi.org/10.3390/pharmaceutics17020273
APA StyleJeon, S. H., Kim, M. C., Lee, H., Oh, J.-H., Kim, H. S., Lee, H., Park, T., & Lee, Y.-J. (2025). A Sequential Ultrafiltration Method to Enhance the Accuracy and Throughput in Plasma Protein Binding Tests. Pharmaceutics, 17(2), 273. https://doi.org/10.3390/pharmaceutics17020273