
Integrating In Vitro Dissolution and Physiologically Based Pharmacokinetic Modeling for Generic Drug Development: Evaluation of Amorphous Solid Dispersion Formulations for Tacrolimus



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Formulation Preparation
2.3. In Vitro Dissolution Testing
2.4. Clinical Data
2.5. PBPK Model Building
2.5.1. Software Tools and PBPK Model Development
2.5.2. In Vitro–In Vivo Extrapolation Approach
2.5.3. Model Performance
3. Results and Discussion
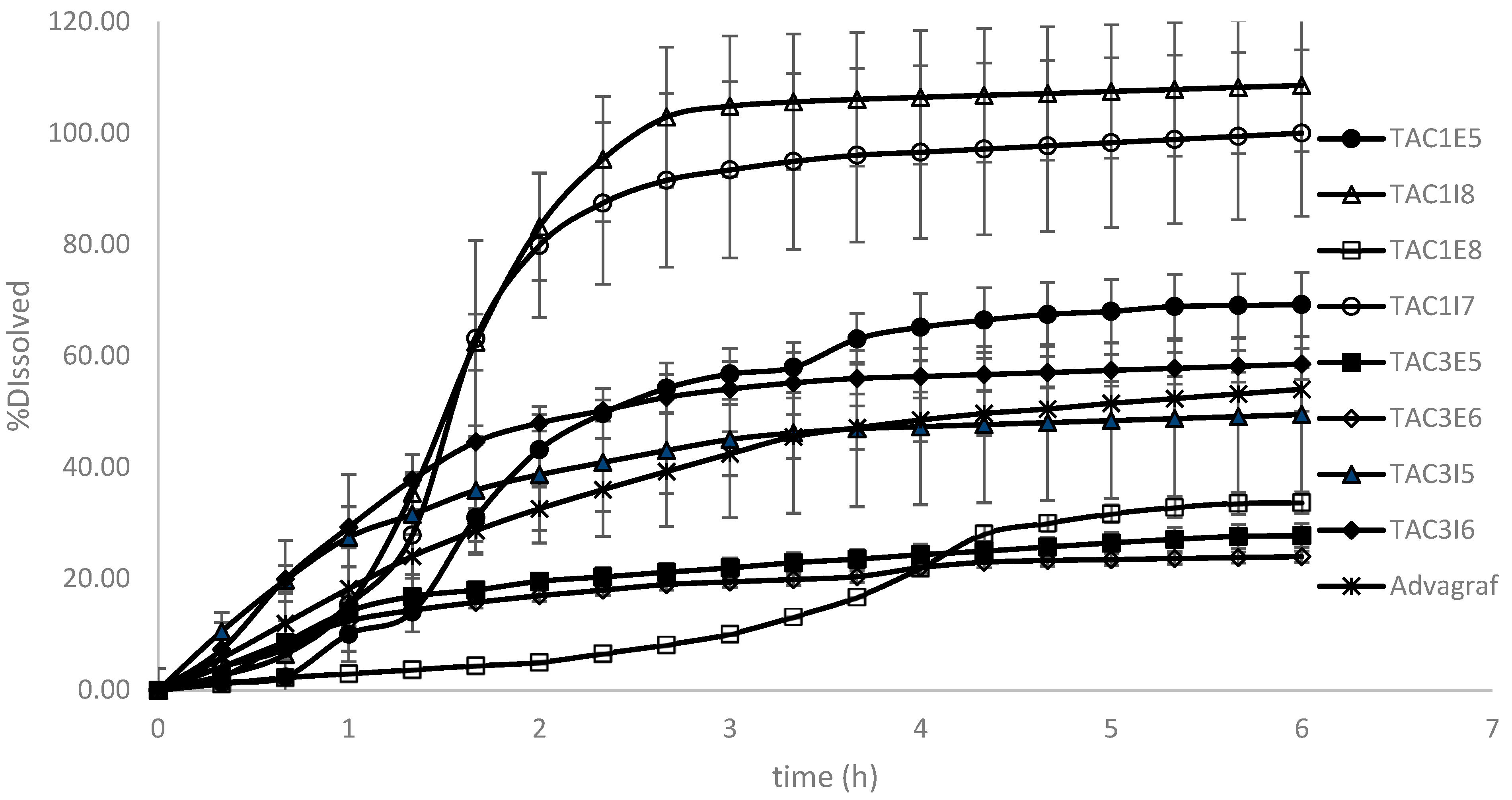
3.1. Dissolution Modeling
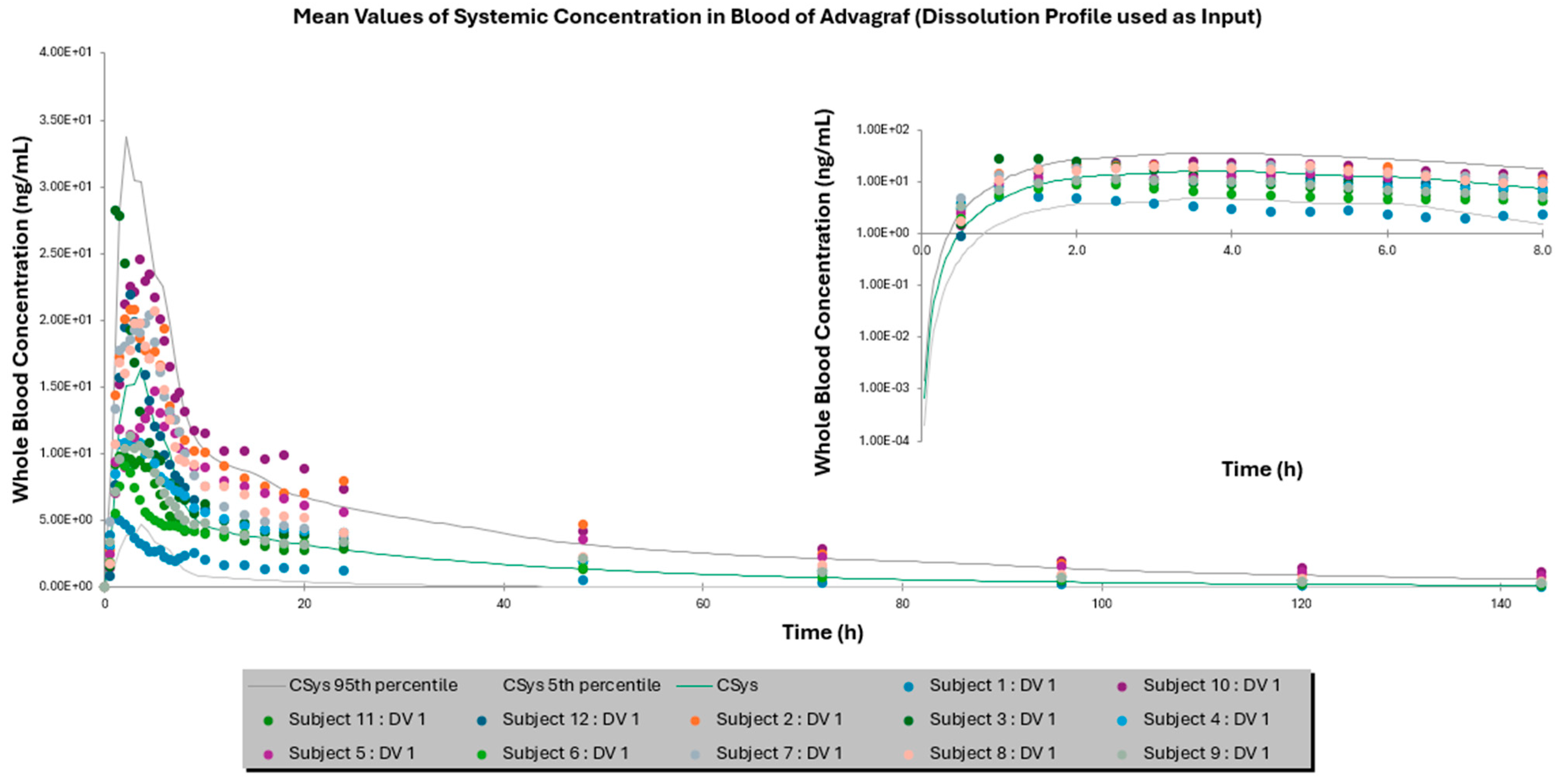
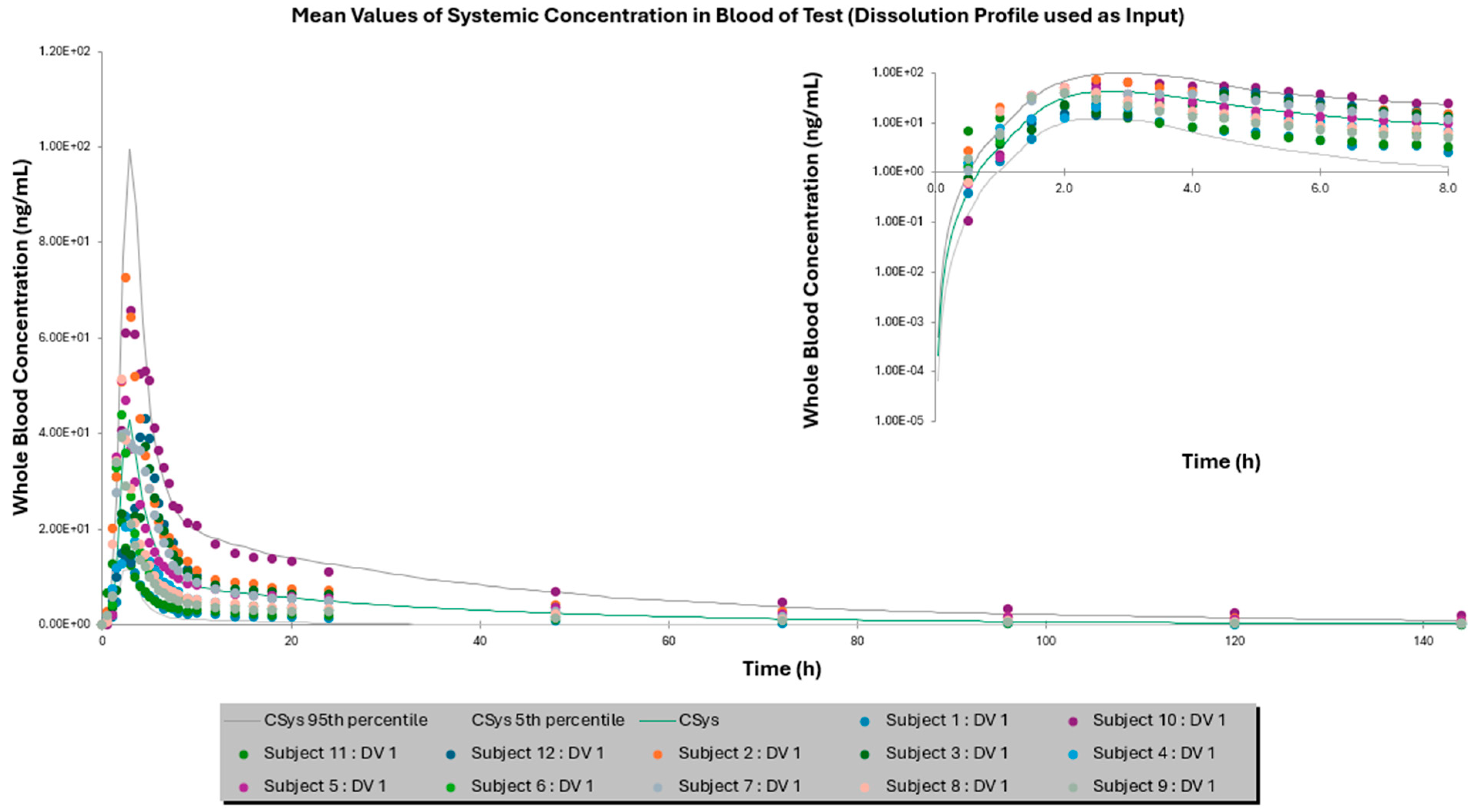
3.2. Comparison of IVIVE Predictions to the Clinical Study
3.3. IVIVE to Explore Potential Test Product Candidates for Bioequivalence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaur, N.; Narang, A.; Bansal, A.K. Use of Biorelevant Dissolution and PBPK Modeling to Predict Oral Drug Absorption. Eur. J. Pharm. Biopharm. 2018, 129, 222–246. [Google Scholar] [CrossRef]
- Ghate, V.M.; Chaudhari, P.; Lewis, S.A. Physiologically Based Pharmacokinetic (PBPK) Modelling for In Vitro-In Vivo Extrapolation: Emphasis on the Use of Dissolution Data. Dissolution Technol. 2019, 26, 18–27. [Google Scholar] [CrossRef]
- Zhang, F.; Zhou, Y.; Wu, N.; Jia, R.; Liu, A.; Liu, B.; Zhou, Z.; Hu, H.; Han, Z.; Ye, X.; et al. In Silico Prediction of Bioequivalence of Isosorbide Mononitrate Tablets with Different Dissolution Profiles Using PBPK Modeling and Simulation. Eur. J. Pharm. Sci. 2021, 157, 105618. [Google Scholar] [CrossRef]
- Kato, T.; Nakagawa, H.; Mikkaichi, T.; Miyano, T.; Matsumoto, Y.; Ando, S. Establishment of a Clinically Relevant Specification for Dissolution Testing Using Physiologically Based Pharmacokinetic (PBPK) Modeling Approaches. Eur. J. Pharm. Biopharm. 2020, 151, 45–52. [Google Scholar] [CrossRef]
- Kambayashi, A.; Kiyota, T.; Fujiwara, M.; Dressman, J.B. PBPK Modeling Coupled with Biorelevant Dissolution to Forecast the Oral Performance of Amorphous Solid Dispersion Formulations. Eur. J. Pharm. Sci. 2019, 135, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Fang, M.; Zhang, Q.; Li, X.; Zhang, T. Evaluating the Bioequivalence of Metronidazole Tablets and Analyzing the Effect of in Vitro Dissolution on in Vivo Absorption Based on PBPK Modeling. Drug Dev. Ind. Pharm. 2019, 45, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Thakur, P.S.; Shete, G.; Gangwal, R.; Sangamwar, A.T.; Bansal, A.K. Understanding the Oral Absorption of Irbesartan Using Biorelevant Dissolution Testing and PBPK Modeling. AAPS PharmSciTech 2020, 21, 102. [Google Scholar] [CrossRef] [PubMed]
- Segregur, D.; Barker, R.; Mann, J.; Moir, A.; Karlsson, E.M.; Turner, D.B.; Arora, S.; Dressman, J. Evaluating the Impact of Acid-Reducing Agents on Drug Absorption Using Biorelevant in Vitro Tools and PBPK Modeling—Case Example Dipyridamole. Eur. J. Pharm. Sci. 2021, 160, 105750. [Google Scholar] [CrossRef]
- Chakraborty, S.; Yadav, L.; Aggarwal, D. Prediction of in Vivo Drug Performance Using in Vitro Dissolution Coupled with STELLA: A Study with Selected Drug Products. Drug Dev. Ind. Pharm. 2015, 41, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- Purohit, H.S.; Trasi, N.S.; Sun, D.D.; Chow, E.C.Y.; Wen, H.; Zhang, X.; Gao, Y.; Taylor, L.S. Investigating the Impact of Drug Crystallinity in Amorphous Tacrolimus Capsules on Pharmacokinetics and Bioequivalence Using Discriminatory In Vitro Dissolution Testing and Physiologically Based Pharmacokinetic Modeling and Simulation. J. Pharm. Sci. 2018, 107, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.F.; Ashtikar, M.; Kojima, R.; Yoshida, T.; Kaihara, M.; Tajiri, T.; Shanehsazzadeh, S.; Modh, H.; Wacker, M.G. Predicting Drug Release and Degradation Kinetics of Long-Acting Microsphere Formulations of Tacrolimus for Subcutaneous Injection. J. Control. Release 2021, 329, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Houston, J.B.; Galetin, A. Physiologically Based Pharmacokinetic Modeling of Intestinal First-Pass Metabolism of CYP3A Substrates with High Intestinal Extraction. Drug Metab. Dispos. 2011, 39, 1633–1642. [Google Scholar] [CrossRef]
- Pei, L.; Li, R.; Zhou, H.; Du, W.; Gu, Y.; Jiang, Y.; Wang, Y.; Chen, X.; Sun, J.; Zhu, J. A Physiologically Based Pharmacokinetic Approach to Recommend an Individual Dose of Tacrolimus in Adult Heart Transplant Recipients. Pharmaceutics 2023, 15, 2580. [Google Scholar] [CrossRef]
- Emoto, C.; Johnson, T.N.; Hahn, D.; Christians, U.; Alloway, R.R.; Vinks, A.A.; Fukuda, T. A Theoretical Physiologically-Based Pharmacokinetic Approach to Ascertain Covariates Explaining the Large Interpatient Variability in Tacrolimus Disposition. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 273–284. [Google Scholar] [CrossRef]
- Van Der Veken, M.; Brouwers, J.; Ozbey, A.C.; Umehara, K.; Stillhart, C.; Knops, N.; Augustijns, P.; Parrott, N.J. Investigating Tacrolimus Disposition in Paediatric Patients with a Physiologically Based Pharmacokinetic Model Incorporating CYP3A4 Ontogeny, Mechanistic Absorption and Red Blood Cell Binding. Pharmaceutics 2023, 15, 2231. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Bu, F.; Wang, Q.; Li, M.; Lin, J.; Tang, Z.; Mak, W.Y.; Zhuang, X.; Zhu, X.; Lin, H.-S.; et al. Examination of the Impact of CYP3A4/5 on Drug–Drug Interaction between Schizandrol A/Schizandrol B and Tacrolimus (FK-506): A Physiologically Based Pharmacokinetic Modeling Approach. Int. J. Mol. Sci. 2022, 23, 4485. [Google Scholar] [CrossRef] [PubMed]
- Tsakiridou, G.; O’Dwyer, P.J.; Margaritis, A.; Box, K.J.; Vertzoni, M.; Kalantzi, L.; Reppas, C. On the Usefulness of Four in Vitro Methodologies in Screening for Product Related Differences in Tacrolimus Exposure after Oral Administration of Amorphous Solid Dispersions with Modified Release Characteristics in the Fasted State. J. Drug Deliv. Sci. Technol. 2022, 69, 102990. [Google Scholar] [CrossRef]
- Tsakiridou, G.; Reppas, C.; Kuentz, M.; Kalantzi, L. A Novel Rheological Method to Assess Drug-Polymer Interactions Regarding Miscibility and Crystallization of Drug in Amorphous Solid Dispersions for Oral Drug Delivery. Pharmaceutics 2019, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, C.; Andreas, C.J.; Vertzoni, M.; Dressman, J.; Reppas, C. In-Vitro Simulation of Luminal Conditions for Evaluation of Performance of Oral Drug Products: Choosing the Appropriate Test Media. Eur. J. Pharm. Biopharm. 2015, 93, 173–182. [Google Scholar] [CrossRef]
- Reppas, C.; Vrettos, N.-N.; Dressman, J.; Andreas, C.J.; Miyaji, Y.; Brown, J.; Etherson, K.; Hanley, S.; Karkossa, F.; Karlsson, E.; et al. Dissolution Testing of Modified Release Products with Biorelevant Media: An OrBiTo Ring Study Using the USP Apparatus III and IV. Eur. J. Pharm. Biopharm. 2020, 156, 40–49. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, M.; Luo, M.; Cai, T. Advances in the Development of Amorphous Solid Dispersions: The Role of Polymeric Carriers. Asian J. Pharm. Sci. 2023, 18, 100834. [Google Scholar] [CrossRef] [PubMed]
- Everaerts, M.; Cools, L.; Adriaensens, P.; Reekmans, G.; Baatsen, P.; Van Den Mooter, G. Investigating the Potential of Ethyl Cellulose and a Porosity-Increasing Agent as a Carrier System for the Formulation of Amorphous Solid Dispersions. Mol. Pharm. 2022, 19, 2712–2724. [Google Scholar] [CrossRef]
- Chaudhari, S.P.; Dugar, R.P. Application of Surfactants in Solid Dispersion Technology for Improving Solubility of Poorly Water Soluble Drugs. J. Drug Deliv. Sci. Technol. 2017, 41, 68–77. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of Poorly Water-Soluble Drugs for Oral Administration: Physicochemical and Physiological Issues and the Lipid Formulation Classification System. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Jusko, W.J.; Piekoszewski, W.; Klintmalm, G.B.; Shaefer, M.S.; Hebert, M.F.; Piergies, A.A.; Lee, C.C.; Schechter, P.; Mekki, Q.A. Pharmacokinetics of Tacrolimus in Liver Transplant Patients*. Clin. Pharmacol. Ther. 1995, 57, 281–290. [Google Scholar] [CrossRef]
- Zahir, H.; Nand, R.A.; Brown, K.F.; Tattam, B.N.; McLachlan, A.J. Validation of Methods to Study the Distribution and Protein Binding of Tacrolimus in Human Blood. J. Pharmacol. Toxicol. Methods 2001, 46, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Ohike, A.; Ibuki, R.; Amidon, G.L.; Yamashita, S. Tacrolimus Is a Class II Low-Solubility High-Permeability Drug: The Effect of P-Glycoprotein Efflux on Regional Permeability of Tacrolimus in Rats. J. Pharm. Sci. 2002, 91, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Bekersky, I.; Dressler, D.; Alak, A.; Boswell, G.W.; Mekki, Q.A. Comparative Tacrolimus Pharmacokinetics: Normal versus Mildly Hepatically Impaired Subjects. J. Clin. Pharmacol. 2001, 41, 628–635. [Google Scholar] [CrossRef]
- Dai, Y.; Hebert, M.F.; Isoherranen, N.; Davis, C.L.; Marsh, C.; Shen, D.D.; Thummel, K.E. Effect of CYP3A5 polymorphism on tacrolimus metabolic clearance in vitro. Drug Metab. Dispos. 2006, 34, 836–847. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | Polymer Type | Surfactant Type (%) | Surfactant Incorporation Mode |
|---|---|---|---|
| TAC1E5 | Eudragit S100 | Solutol (0.5%) | extra-granularly |
| TAC1E8 | Eudragit S100 | Solutol (3.5%) | extra-granularly |
| TAC1I8 (Test) * | Eudragit S100 | Solutol (3.5%) | intra-granularly |
| TAC1I7 | Eudragit S100 | Solutol (0.5%) | intra-granularly |
| TAC3E5 | Ethylcellulose | Soluplus (0.5%) | extra-granularly |
| TAC3E6 | Ethylcellulose | Soluplus (3.5%) | extra-granularly |
| TAC3I5 | Ethylcellulose | Soluplus (0.5%) | intra-granularly |
| TAC3I6 | Ethylcellulose | Soluplus (3.5%) | intra-granularly |
| Simulated GI Region | Dissolution Medium | Start Time | Duration | Flow Rate (mL/min) |
|---|---|---|---|---|
| Stomach | FaSSGF | 0 | 30 | 8 |
| Proximal gut | FaSSIF-V2 | 30 | 40 | 4 |
| Midgut | FaSSIF-V2 midgut | 70 | 80 | 4 |
| Distal Ileum | SIF Ileum-V2 | 150 | 60 | 4 |
| Ascending Colon | FaSSCoF | 210 | 150 | 4 |
| Advagraf® | Test Formulation | |||||
|---|---|---|---|---|---|---|
| Observed | Simulated with Dissolution Profile | Simulated with DLM | Observed | Simulated with Dissolution Profile | Simulated with DLM * | |
| Mean AUC0–144 h ± SD (ng × h/mL) | 355.87 ± 160.22 | 245.80 ± 177.43 | 378.12 ± 298.15 | 462.97 ± 267.64 | 429.15 ± 380.33 | 457.86 ± 404.33 |
| Fold Error | - | 1.54 | 1.06 | - | 1.08 | 1.01 |
| Mean Cmax ± SD (ng/mL) | 16.46 ± 7.18 | 17.19 ± 11.74 | 13.10 ± 8.27 | 43.23 ± 15.54 | 42.77 ± 28.60 | 37.03 ± 23.746 |
| Fold Error | - | 1.04 | 1.26 | - | 1.01 | 1.17 |
| Mean Tmax ± SD (h) | 2.92 ± 1.38 | 3.05 ± 0.83 | 3.77 ± 0.9 | 2.66 ± 0.96 | 2.83 ± 0.18 | 2.93 ± 0.51 |
| Fold Error | - | 1.04 | 1.29 | - | 1.06 | 1.10 |
| Test Formulation with Constant DLM Scalar in Colon, Equal to 0.5 | Test Formulation with Constant DLM Scalar in Ileum Compartments (I to IV), Equal to 0.05 | |||||
|---|---|---|---|---|---|---|
| Ileum DLM Scalar = 0.005 | Ileum DLM Scalar = 0.05 | Ileum DLM Scalar = 0.5 | Colon DLM Scalar = 0.05 | Colon DLM Scalar = 0.5 | Colon DLM Scalar = 5.0 | |
| Mean AUC0–144 h (ng × h/mL) | 561.26 | 457.86 | 373.52 | 437.36 | 457.86 | 461.22 |
| Mean Cmax (ng/mL) | 37.23 | 37.03 | 39.53 | 30.39 | 37.03 | 38.61 |
| Mean Tmax (h) | 3.81 | 2.93 | 2.18 | 2.82 | 2.93 | 2.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakitsios, E.; Angelerou, M.-F.-G.; Kapralos, I.; Tsakiridou, G.; Kalantzi, L.; Dokoumetzidis, A. Integrating In Vitro Dissolution and Physiologically Based Pharmacokinetic Modeling for Generic Drug Development: Evaluation of Amorphous Solid Dispersion Formulations for Tacrolimus. Pharmaceutics 2025, 17, 227. https://doi.org/10.3390/pharmaceutics17020227
Karakitsios E, Angelerou M-F-G, Kapralos I, Tsakiridou G, Kalantzi L, Dokoumetzidis A. Integrating In Vitro Dissolution and Physiologically Based Pharmacokinetic Modeling for Generic Drug Development: Evaluation of Amorphous Solid Dispersion Formulations for Tacrolimus. Pharmaceutics. 2025; 17(2):227. https://doi.org/10.3390/pharmaceutics17020227
Chicago/Turabian StyleKarakitsios, Evangelos, Maria-Faidra-Galini Angelerou, Iasonas Kapralos, Georgia Tsakiridou, Lida Kalantzi, and Aristides Dokoumetzidis. 2025. "Integrating In Vitro Dissolution and Physiologically Based Pharmacokinetic Modeling for Generic Drug Development: Evaluation of Amorphous Solid Dispersion Formulations for Tacrolimus" Pharmaceutics 17, no. 2: 227. https://doi.org/10.3390/pharmaceutics17020227
APA StyleKarakitsios, E., Angelerou, M.-F.-G., Kapralos, I., Tsakiridou, G., Kalantzi, L., & Dokoumetzidis, A. (2025). Integrating In Vitro Dissolution and Physiologically Based Pharmacokinetic Modeling for Generic Drug Development: Evaluation of Amorphous Solid Dispersion Formulations for Tacrolimus. Pharmaceutics, 17(2), 227. https://doi.org/10.3390/pharmaceutics17020227