

Preparing a Liposome-Aided Drug Delivery System: The Entrapment and Release Profiles of Doxorubicin and 9-(N-Piperazinyl)-5-methyl-12(H)-quino [3,4-b][1,4]benzothiazinium Chloride with Human Serum Albumin

 , , ,
, , ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Liposome Preparation
2.3. Solutions and Sample Preparation
2.4. UV/Vis Measurements
2.5. Drug Release Versus Mathematical Modeling
2.6. Encapsulation Efficiency
2.7. Zeta Potential and Particle Size Measurements
2.8. Statistical Analysis
3. Results and Discussion
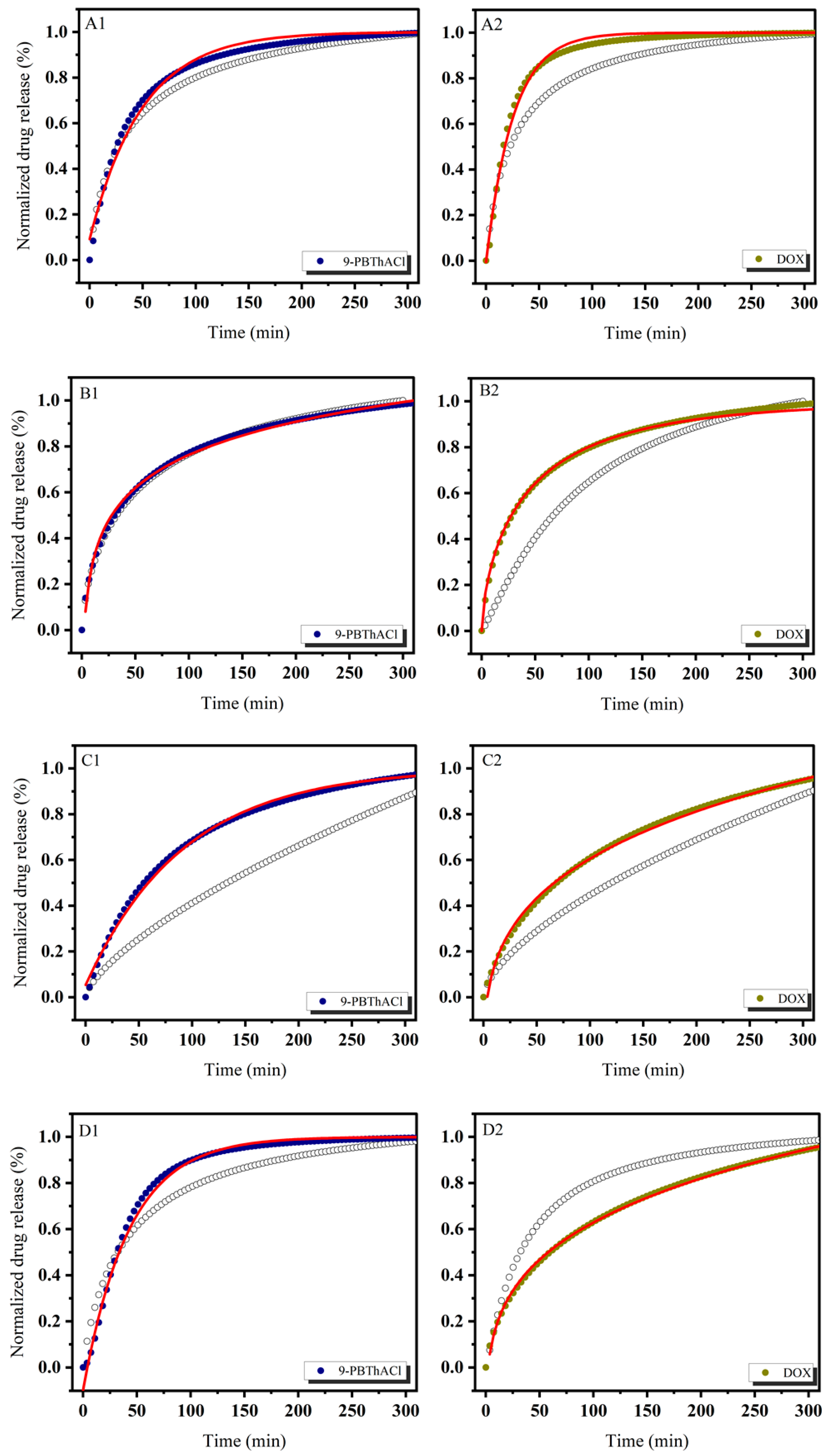
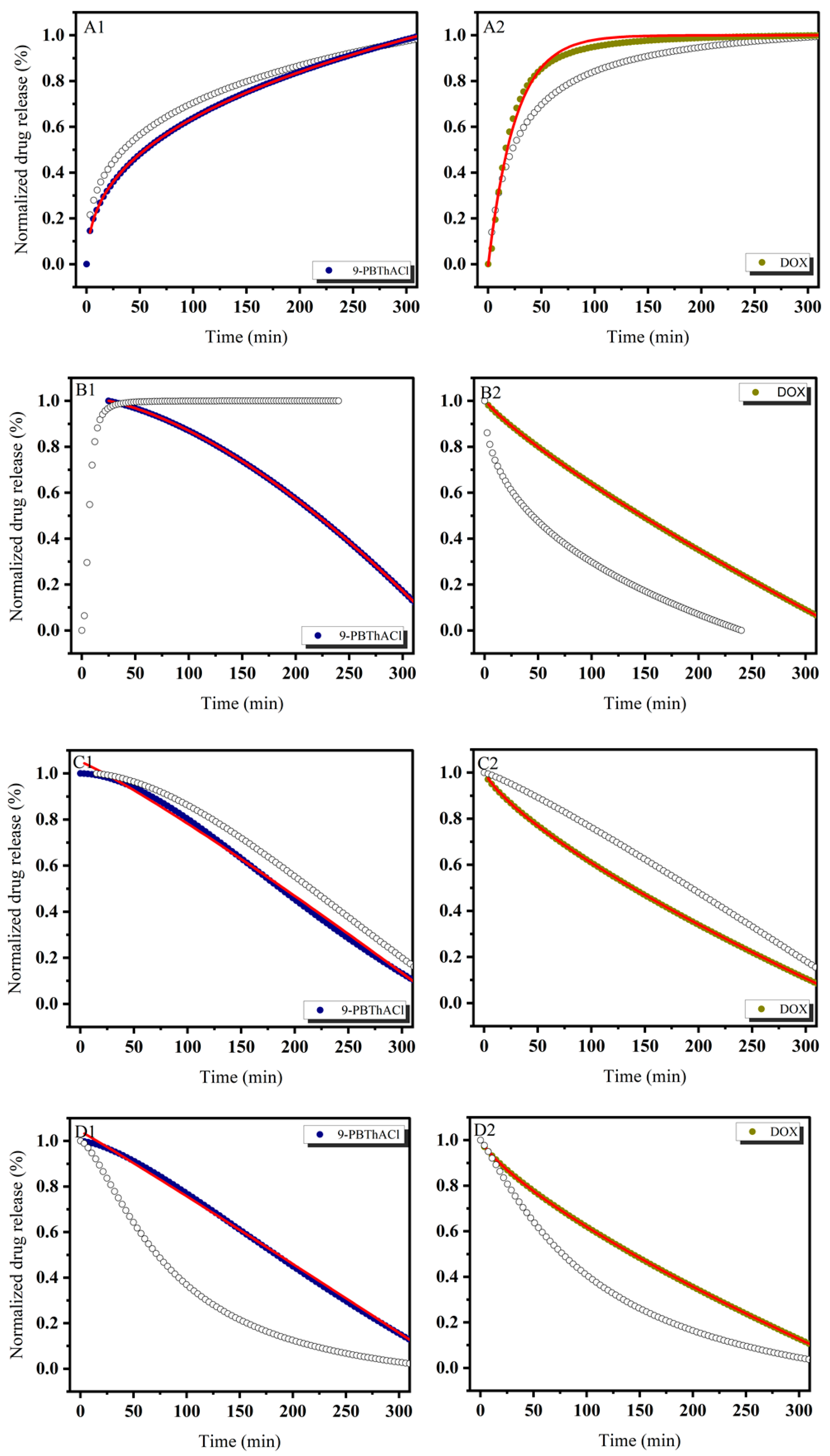
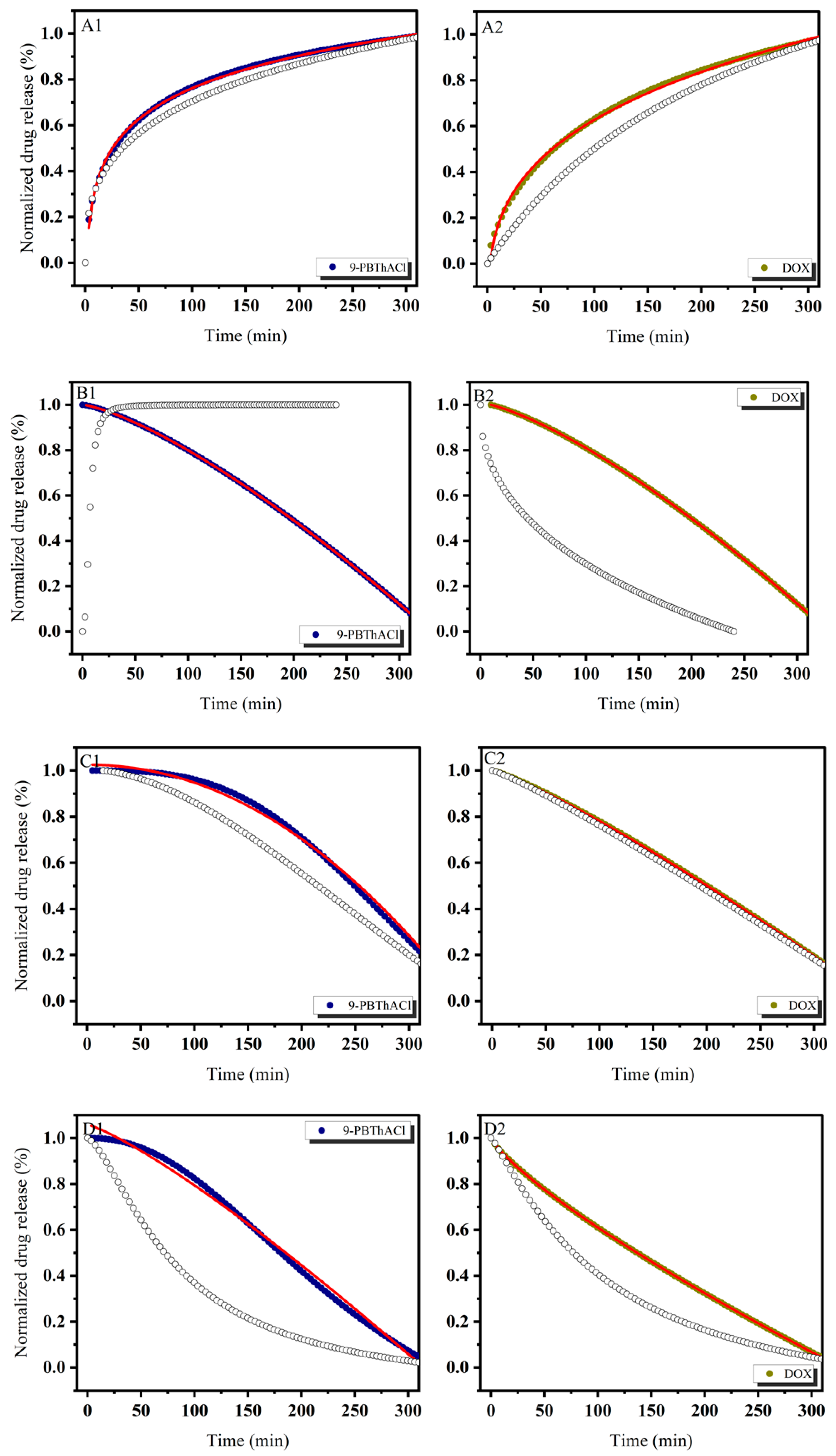
3.1. In Vitro Drug Release Evaluation
3.2. Characteristics of Liposomes
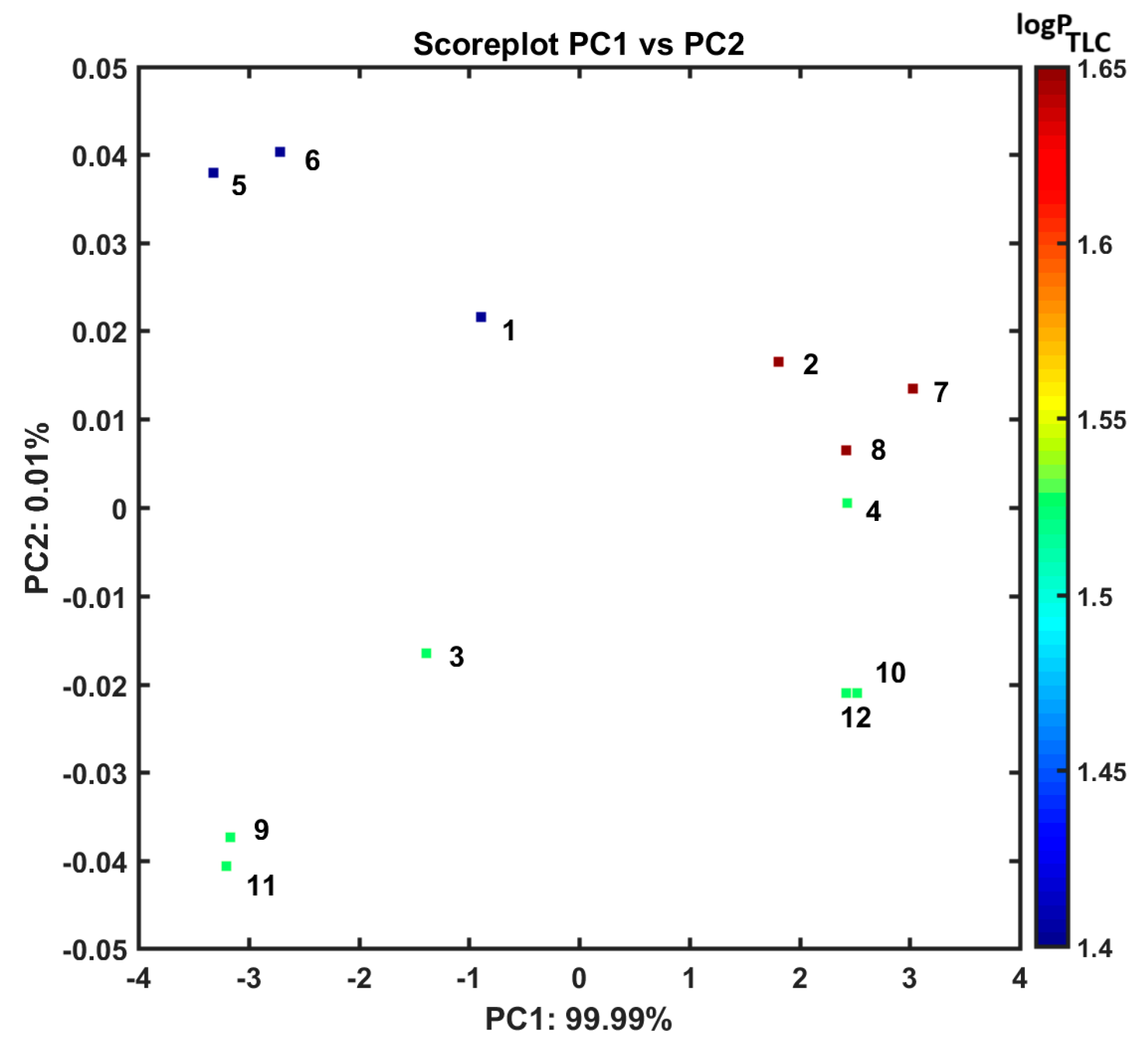
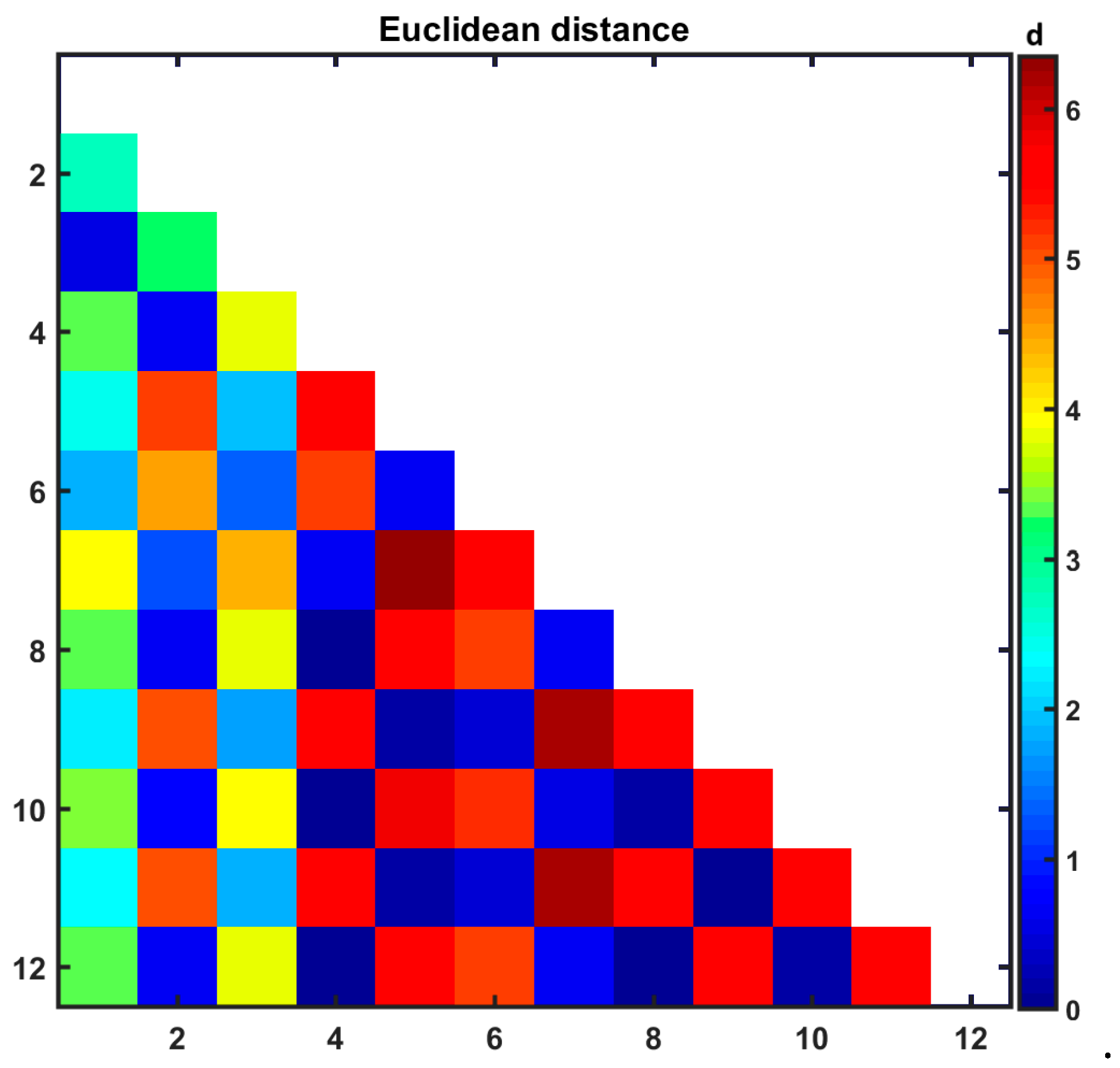
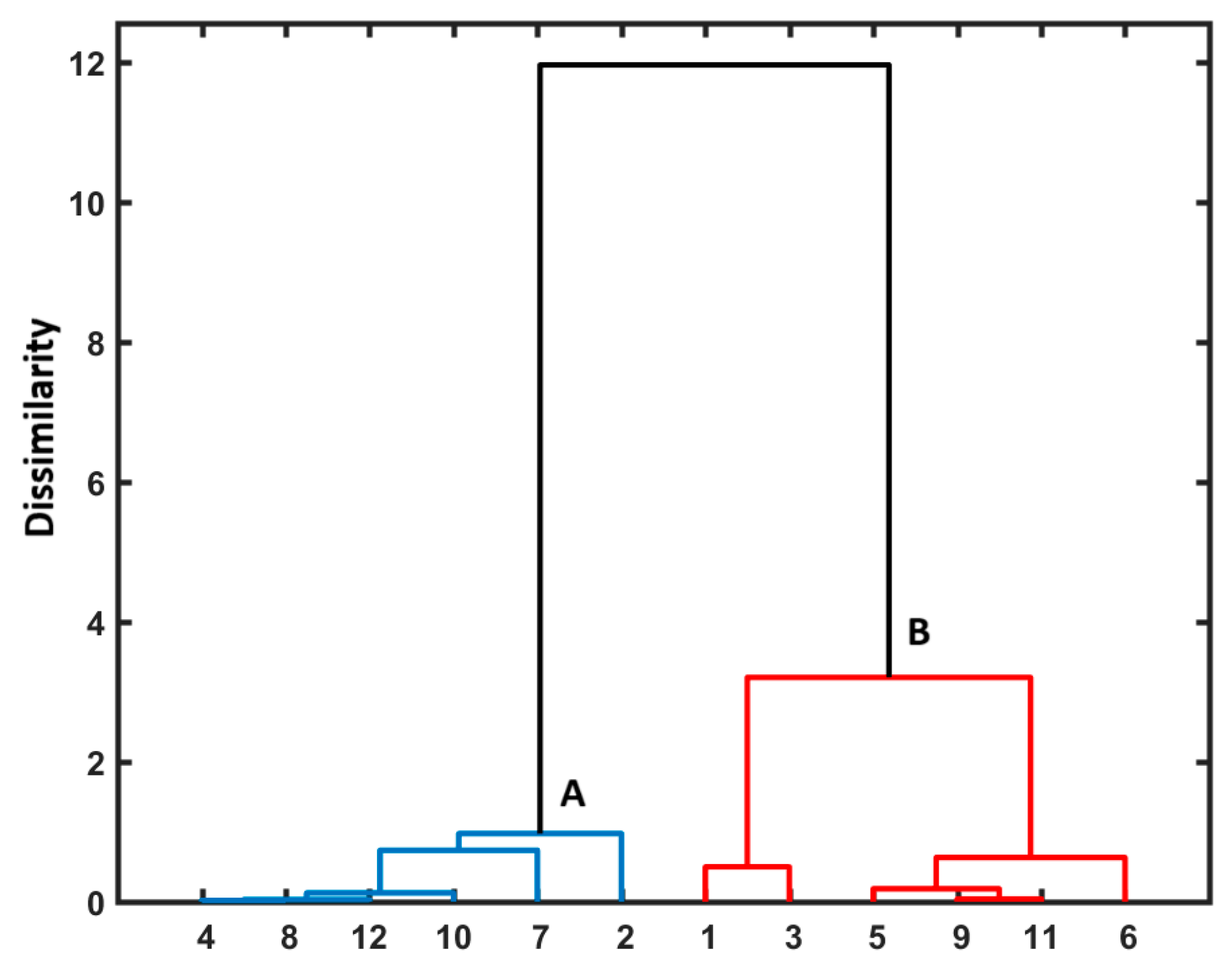
3.3. Similarity-Guided Drug Release Evaluation
3.4. One-Factor Analysis of Variance (ANOVA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, C.; Zhang, D.; Pan, Y.; Chen, B. Human serum albumin based nanodrug delivery systems: Recent advances and future perspective. Polymers 2023, 15, 3354. [Google Scholar] [CrossRef]
- Gatto, M.S.; Johnson, M.P.; Najahi-Missaoui, W. Targeted liposomal drug delivery: Overview of the current applications and challenges. Life 2024, 14, 672. [Google Scholar] [CrossRef] [PubMed]
- Nsairat, H.; Ibrahim, A.A.; Jaber, A.M.; Abdelghany, S.; Atwan, R.; Shalan, N.; Abdelnabi, H.; Odeh, F.; El-Tanani, M.; Alshaer, W. Liposome bilayer stability: Emphasis on cholesterol and its alternatives. J. Liposome Res. 2024, 34, 178–202. [Google Scholar] [CrossRef] [PubMed]
- Fulton, M.D.; Najahi-Missaoui, W. Liposomes in cancer therapy: How did we start and where are we now. Int. J. Mol. Sci. 2023, 24, 6615. [Google Scholar] [CrossRef] [PubMed]
- Atre, P.; Rizvi, A.A.S. A brief overview of quality by design approach for developing pharmaceutical liposomes as nano-sized parenteral drug delivery systems. RSC Pharm. 2024, 1, 675–688. [Google Scholar] [CrossRef]
- Zahednezhad, F.; Allahyari, S.; Sarfraz, M.; Zakeri-Milani, P.; Feyzizadeh, M.; Valizadeh, H. Liposomal drug delivery systems for organ-specific cancer targeting: Early promises, subsequent problems, and recent breakthroughs. Expert Opin. Drug Deliv. 2024, 21, 1363–1384. [Google Scholar] [CrossRef]
- Eugster, R.; Luciani, P. Liposomes: Bridging the gap from lab to pharmaceuticals. Curr. Opin. Colloid Interface Sci. 2025, 75, 101875. [Google Scholar] [CrossRef]
- Abboud, H.A.; Zelkó, R.; Kazsoki, A. A systematic review of liposomal nanofibrous scaffolds as a drug delivery system: A decade of progress in controlled release and therapeutic efficacy. Drug Deliv. 2025, 32, 2445259. [Google Scholar] [CrossRef]
- Tao, H.; Wang, R.; Sheng, W.; Zhen, Y. The development of human serum albumin-based drugs and relevant fusion proteins for cancer therapy. Int. J. Biol. Macromol. 2021, 187, 24–34. [Google Scholar] [CrossRef]
- Al-Harthi, S.; Lachowicz, J.I.; Nowakowski, M.E.; Jaremko, M.; Jaremko, L. Towards the functional high-resolution coordination chemistry of blood plasma human serum albumin. J. Inorg. Biochem. 2019, 198, 110716. [Google Scholar] [CrossRef]
- Bhushan, B.; Khanadeev, V.; Khlebtsov, B.; Khlebtsov, N.; Gopinath, P. Impact of albumin-based approaches in nanomedicine: Imaging, targeting and drug delivery. Adv. Colloid Interface Sci. 2017, 246, 13–39. [Google Scholar] [CrossRef] [PubMed]
- di Masi, A. Human serum albumin: From molecular aspects to biotechnological applications. Int. J. Mol. Sci. 2023, 24, 4081. [Google Scholar] [CrossRef] [PubMed]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar] [CrossRef] [PubMed]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef]
- Olivieri, J.R.; Craievich, A.F. The subdomain structure of human serum albumin in solution under different pH conditions studied by small angle X-ray scattering. Eur. Biophys. J. 1995, 24, 77–84. [Google Scholar]
- Ferrer, M.L.; Duchowicz, R.; Carrasco, B.; de la Torre, J.G.; Acuna, A.U. The conformation of serum albumin in solution: A combined phosphorescence depolarization-hydrodynamic modelling study. Biophys. J. 2001, 80, 2422–2430. [Google Scholar] [CrossRef]
- Zeeshan, F.; Madheswaran, T.; Panneerselvam, J.; Taliyan, R.; Kesharwani, P. Human serum albumin as multifunctional nanocarrier for cancer therapy. J. Pharm. Sci. 2021, 110, 3111–3117. [Google Scholar] [CrossRef]
- Yamasaki, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Albumin-drug interaction and its clinical implication. Biochim. Biophys. Acta 2013, 1830, 5435–5443. [Google Scholar] [CrossRef]
- Dreis, S.; Rothweiler, F.; Michaelis, M.; Cinatl, J., Jr.; Kreuter, J.; Langer, K. Preparation, characterisation and maintenance of drug efficacy of doxorubicin-loaded human serum albumin (HSA) nanoparticles. Int. J. Pharm. 2007, 341, 207–214. [Google Scholar] [CrossRef]
- Onafuye, H.; Pieper, S.; Mulac, D.; Cinatl, J., Jr.; Wass, M.N.; Langer, K.; Michaelis, M. Doxorubicin-loaded human serum albumin nanoparticles overcome transporter-mediated drug resistance in drug-adapted cancer cells. Beilstein J. Nanotechnol. 2019, 10, 1707–1715. [Google Scholar] [CrossRef]
- Ivanov, A.I.; Christodoulou, J.; Parkinson, J.A.; Barnham, K.J.; Tucker, A.; Woodrow, J.; Sadler, P.J. Cisplatin binding sites on human albumin. J. Biol. Chem. 1998, 273, 14721–14730. [Google Scholar] [CrossRef] [PubMed]
- Park, C.R.; Kim, H.Y.; Song, M.G.; Lee, Y.S.; Youn, H.; Chung, J.K.; Cheon, G.J.; Kang, K.W. Efficacy and safety of human serum albumin-cisplatin complex in U87MG xenograft mouse models. Int. J. Mol. Sci. 2020, 21, 7932. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Sun, Y.; Li, Y.; Hao, F.; Qiu, P.; Teng, L.; Xie, J.; Gao, Y. Docetaxel-loaded human serum albumin (HSA) nanoparticles: Synthesis, characterization, and evaluation. Biomed. Eng. Online 2019, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, M.; Wang, H.; Zhou, J.; Chen, J. Multifunctional human serum albumin fusion protein as a docetaxel nanocarrier for chemo-photothermal synergetic therapy of ovarian cancer. ACS Appl. Mater. Interfaces 2022, 14, 19907–19917. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, M.J.; Soon-Shiong, P.; Desai, N. Protein nanoparticles as drug carriers in clinical medicine. Adv. Drug Deliv. Rev. 2008, 60, 876–885. [Google Scholar] [CrossRef]
- Jackson, D.V.; Nichols, A.P.; Bender, R.A. Interaction of albumin and vincristine with a human lymphoblastic leukemia cell line in vitro. Cancer Biochem. Biophys. 1980, 4, 133–136. [Google Scholar]
- Almond, B.A.; Hadba, A.R.; Freeman, S.T.; Cuevas, B.J.; York, A.M.; Detrisac, C.J.; Goldberg, E.P. Efficacy of mitoxantrone-loaded albumin microspheres for intratumoral chemotherapy of breast cancer. J. Control. Release 2003, 91, 147–155. [Google Scholar] [CrossRef]
- Wang, W.; Sun, Q.; Gan, N.; Zhai, Y.; Xiang, H.; Li, H. Characterizing the interaction between methyl ferulate and human serum albumin by saturation transfer difference NMR. RSC Adv. 2020, 10, 32999–33009. [Google Scholar] [CrossRef]
- Xie, J.; Wang, J.; Niu, G.; Huang, J.; Chen, K.; Li, X.; Chen, X. Human serum albumin coated iron oxide nanoparticles for efficient cell labeling. Chem. Commun. 2010, 46, 433–435. [Google Scholar] [CrossRef]
- Bychkova, A.V.; Yakunina, M.N.; Lopukhova, M.V.; Degtyarev, Y.N.; Motyakin, M.V.; Pokrovsky, V.S.; Kovarski, A.L.; Gorobets, M.G.; Retivov, V.M.; Khachatryan, D.S. Albumin-functionalized iron oxide nanoparticles for theranostics: Engineering and long-termin situ imaging. Pharmaceutics 2022, 14, 2771. [Google Scholar] [CrossRef]
- Pentak, D. Evaluation of the physicochemical properties of liposomes as potential carriers of anticancer drugs: Spectroscopic study. J. Nanopart. Res. 2016, 18, 126. [Google Scholar] [CrossRef]
- Pentak, D.; Ploch-Jankowska, A.; Zięba, A.; Kozik, V. The advances and challenges of liposome-assisted drug release in the presence of serum albumin molecules: The influence of surrounding pH. Materials 2022, 15, 1586. [Google Scholar] [CrossRef] [PubMed]
- Spada, A.; Emami, J.; Tuszynski, J.A.; Lavasanifar, A. The uniqueness of albumin as a carrier in nanodrug delivery. Mol. Pharm. 2021, 18, 1862–1894. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, M.-K.; Song, I.-S. Recent advances in doxorubicin formulation to enhance pharmacokinetics and tumor targeting. Pharmaceuticals 2023, 16, 802. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Das, A.; Chakraborty, A. Interaction of human serum albumin with liposomes of saturated and unsaturated lipids with different phase transition temperatures: A spectroscopic investigation by membrane probe PRODAN. RSC Adv. 2014, 4, 14335–14347. [Google Scholar] [CrossRef]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Hershman, D.L.; McBride, R.B.; Eisenberger, A.; Tsai, W.Y.; Grann, V.R.; Jacobson, J.S. Doxorubicin, cardiac risk factors, and cardiac toxicity in elderly patients with diffuse B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol. 2008, 26, 3159–3165. [Google Scholar] [CrossRef]
- Zięba, A.; Maślankiewicz, A.; Suwińska, K. 1-Alkyl-4-arylamino-quinolinium-3-thiolates and 7-alkyl-12H-quino [3,4-b]-1,4-benzothiazinium slats. Eur. J. Org. Chem. 2000, 16, 2947–2953. [Google Scholar] [CrossRef]
- Zięba, A.; Kozik, V.; Suwinska, K.; Kawulok, A.; Pluta, T.; Jampilek, J.; Bak, A. Synthesis and structure of 5-methyl-9-(trifluoromethyl)-12H-quino [3,4-b][1,4]benzothiazinium chloride as anticancer agent. Molecules 2024, 29, 4337. [Google Scholar] [CrossRef]
- Zięba, A.; Latocha, M.; Sochanik, A.; Nycz, A.; Kuśmierz, D. Synthesis and in vitro antiproliferative activity of novel phenylring-substituted5-alkyl-12H-quino [3,4-b][1,4]benzothiazine derivatives. Molecules 2016, 21, 1455. [Google Scholar] [CrossRef] [PubMed]
- Zięba, A.; Sochanik, A.; Szurko, A.; Rams, M.; Mrozek, A.; Cmoch, P. Synthesis and in vitro antiproliferativeactivity of 5-alkyl-12H-quino [3,4-b][1,4]benzothiazinium salts. Eur. J. Med. Chem. 2010, 45, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Szoka, F., Jr.; Papahadjopoulos, D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu. Rev. Biophys. Bioeng. 1980, 9, 467–508. [Google Scholar] [CrossRef]
- Kozik, V.; Pentak, D.; Paździor, M.; Zięba, A.; Bąk, A. From design to study of liposome-driven drug release part 1: Impact of temperature and pH on environment. Int. J. Mol. Sci. 2023, 24, 11686. [Google Scholar] [CrossRef]
- Jin, Y.; Li, M.; Hou, X. Pyrocatechol violet as a marker to characterize liposomal membrane permeability using the chelation and the first-order derivative spectrophotometry. J. Pharm. Biomed. Anal. 2005, 37, 379–382. [Google Scholar] [CrossRef]
- Pentak, D.; Maciazek-Jurczyk, M.; Zawada, Z.H. The role of nanoparticles in the albumin-cytarabine and albumin-methotrexate interactions. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 73, 388–397. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Modeling of diffusion controlled drug delivery. J. Control. Release 2012, 161, 351–362. [Google Scholar] [CrossRef]
- Pentak, D.; Maciazek-Jurczyk, M. Self-assembled nanostructures formed by phospholipids and anticancer drugs. Serum albumin- nanoparticle interactions. J. Mol. Liq. 2016, 224, 1–8. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Alavi, S.E.; Alharthi, S.; Alavi, S.Z.; Raza, A.; Ebrahimi Shahmabadi, H. Bioresponsive drug delivery systems. Drug Discov. Today 2024, 29, 103849. [Google Scholar] [CrossRef]
- Taylor, K.; Morris, R.M. Thermal analysis of phase transition behaviour in liposomes. Thermochim. Acta 1995, 248, 289–3012. [Google Scholar] [CrossRef]
- Smith, M.C.; Crist, R.M.; Clogston, J.D.; Mcneil, S.E. Zeta potential: A case study of cationic, anionic, and neutral liposomes. Anal. Bioanal. Chem. 2017, 409, 5779–5787. [Google Scholar] [CrossRef] [PubMed]
- Herbowski, L.; Gurgul, H.; Staron, W. Experimental determination of the Stern layer thickness at the interface of the human arachnoid membrane and the cerebrospinal fluid. Z. Med. Phys. 2009, 19, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Maja, L.; Željko, K.; Mateja, P. Sustainable technologies for liposome preparation. J. Supercrit. Fluids 2020, 165, 104984. [Google Scholar] [CrossRef]
- Empel, A.; Bak, A.; Kozik, V.; Latocha, M.; Cizek, A.; Jampilek, J.; Suwinska, K.; Sochanik, A.; Zieba, A. Towards property profiling: Synthesis and SAR probing of new tetracyclic diazaphenothiazine analogues. Int. J. Mol. Sci. 2021, 22, 12826. [Google Scholar] [CrossRef]
- Kisiel-Nawrot, E.; Latocha, M.; Bak, A.; Kozik, V.; Jampilek, J.; Zieba, A. Anticancer efficacy of antibacterial quinobenzothiazines. Appl. Sci. 2023, 13, 2886. [Google Scholar] [CrossRef]
- Bertinetto, C.; Engel, J.; Jansen, J. ANOVA simultaneous component analysis: A tutorial review. Anal. Chim. Acta X 2020, 6, 100061. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pentak, D.; Kozik, V.; Zieba, A.; Paździor-Heiske, M.; Szymczyk, A.; Jampilek, J.; Bak, A. Preparing a Liposome-Aided Drug Delivery System: The Entrapment and Release Profiles of Doxorubicin and 9-(N-Piperazinyl)-5-methyl-12(H)-quino [3,4-b][1,4]benzothiazinium Chloride with Human Serum Albumin. Pharmaceutics 2025, 17, 202. https://doi.org/10.3390/pharmaceutics17020202
Pentak D, Kozik V, Zieba A, Paździor-Heiske M, Szymczyk A, Jampilek J, Bak A. Preparing a Liposome-Aided Drug Delivery System: The Entrapment and Release Profiles of Doxorubicin and 9-(N-Piperazinyl)-5-methyl-12(H)-quino [3,4-b][1,4]benzothiazinium Chloride with Human Serum Albumin. Pharmaceutics. 2025; 17(2):202. https://doi.org/10.3390/pharmaceutics17020202
Chicago/Turabian StylePentak, Danuta, Violetta Kozik, Andrzej Zieba, Marlena Paździor-Heiske, Aleksandra Szymczyk, Josef Jampilek, and Andrzej Bak. 2025. "Preparing a Liposome-Aided Drug Delivery System: The Entrapment and Release Profiles of Doxorubicin and 9-(N-Piperazinyl)-5-methyl-12(H)-quino [3,4-b][1,4]benzothiazinium Chloride with Human Serum Albumin" Pharmaceutics 17, no. 2: 202. https://doi.org/10.3390/pharmaceutics17020202
APA StylePentak, D., Kozik, V., Zieba, A., Paździor-Heiske, M., Szymczyk, A., Jampilek, J., & Bak, A. (2025). Preparing a Liposome-Aided Drug Delivery System: The Entrapment and Release Profiles of Doxorubicin and 9-(N-Piperazinyl)-5-methyl-12(H)-quino [3,4-b][1,4]benzothiazinium Chloride with Human Serum Albumin. Pharmaceutics, 17(2), 202. https://doi.org/10.3390/pharmaceutics17020202