
Non-Steroidal Anti-Inflammatory Drugs Are Inhibitors of the Intestinal Proton-Coupled Amino Acid Transporter (PAT1): Ibuprofen and Diclofenac Are Non-Translocated Inhibitors


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Culture
2.2.2. Uptake Experiments in Caco-2 Cells
2.2.3. Xenopus Laevis Oocytes and Two-Electrode Voltage Clamp Measurements
2.2.4. AlphaFold2 Model and Molecular Docking
2.2.5. Data Analysis
2.2.6. Statistical Analysis
3. Results
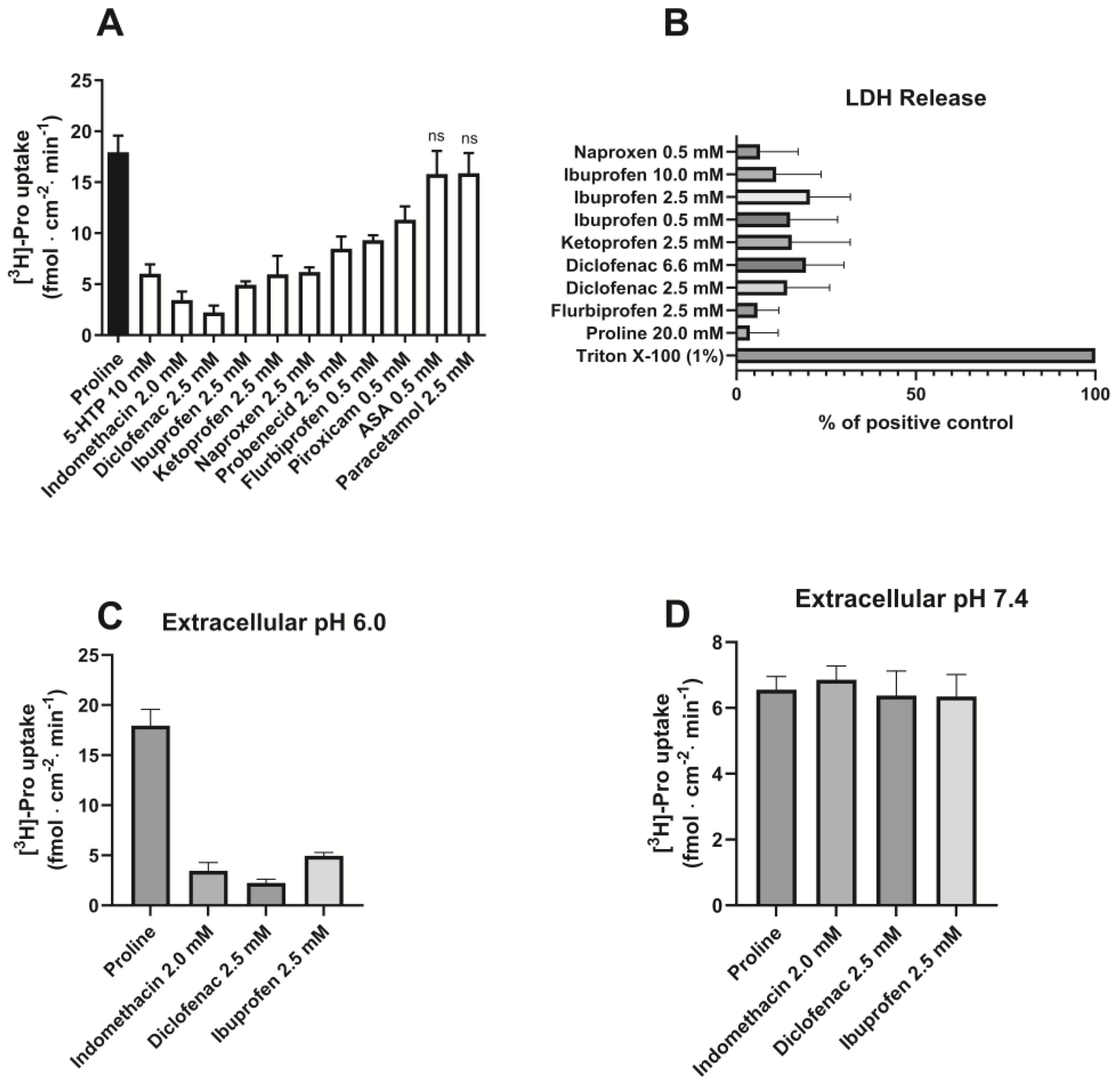
3.1. Various NSAIDs Inhibit PAT1-Mediated Uptake of Proline in Caco-2 Cells
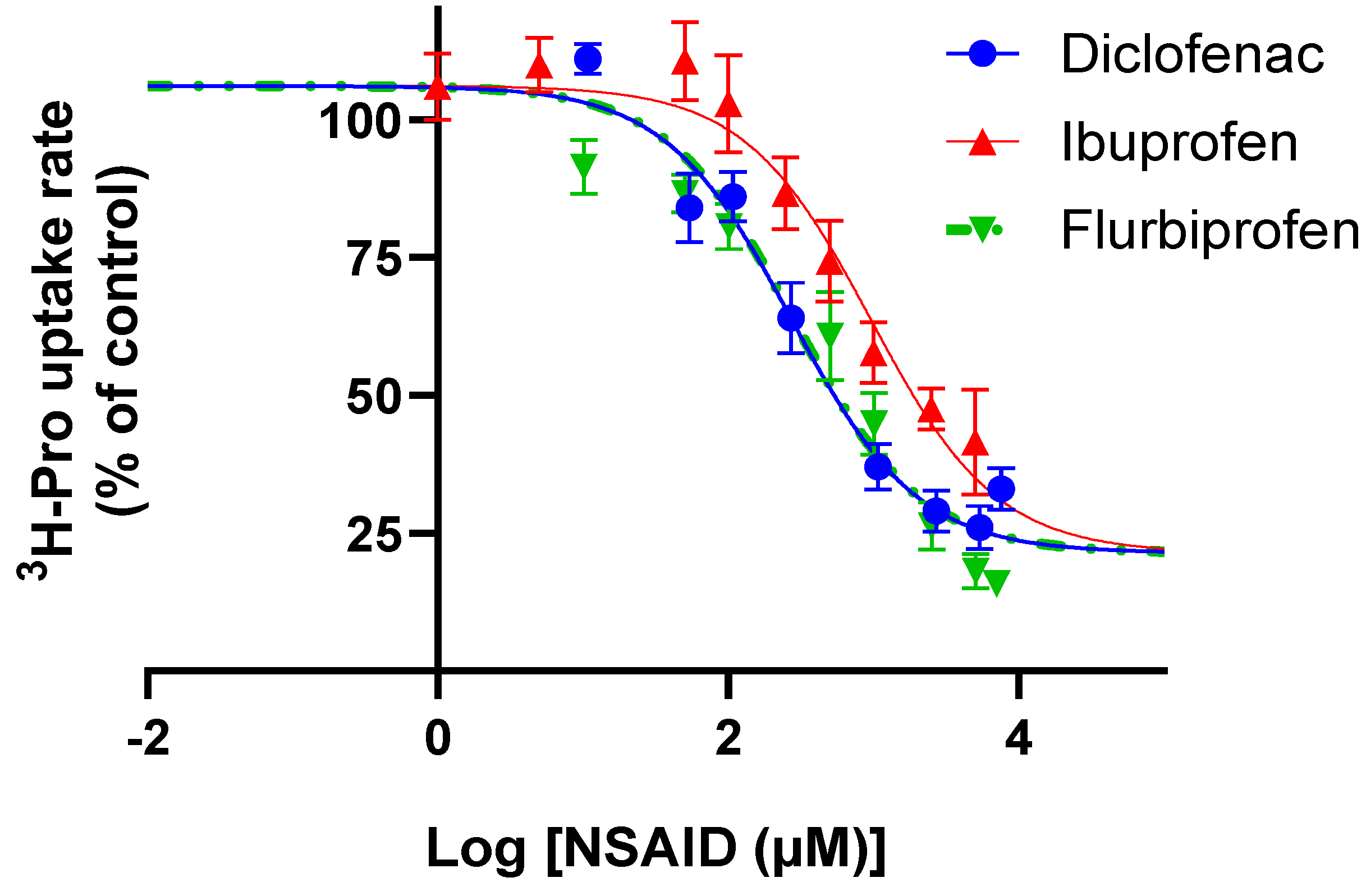
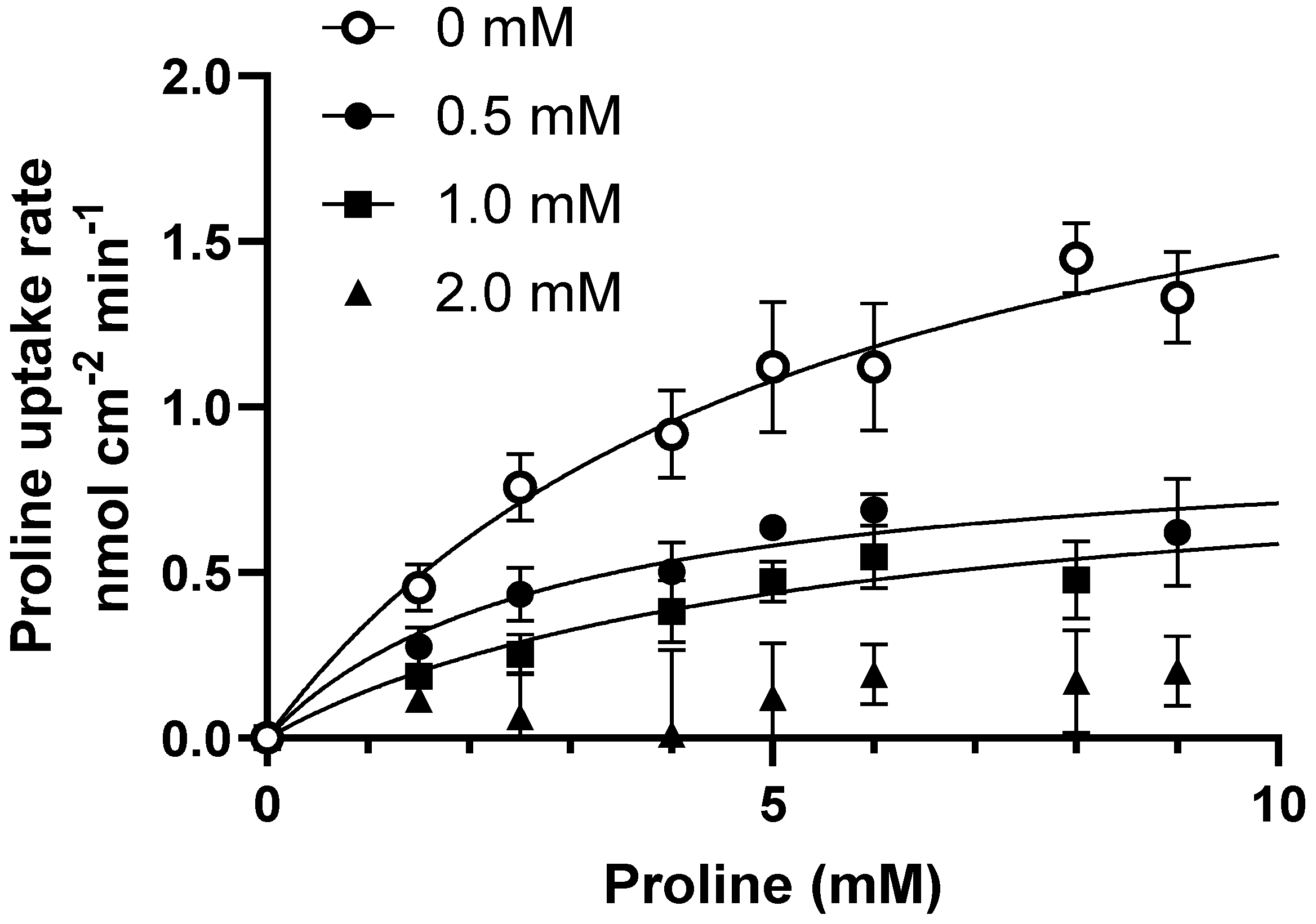
3.2. Ibuprofen, Diclofenac, and Flurbiprofen Concentration-Dependently Inhibit PAT1-Mediated Uptake of Proline
3.3. Ibuprofen and Diclofenac Inhibit Proline-Induced Currents in PAT1-Expressing Oocytes
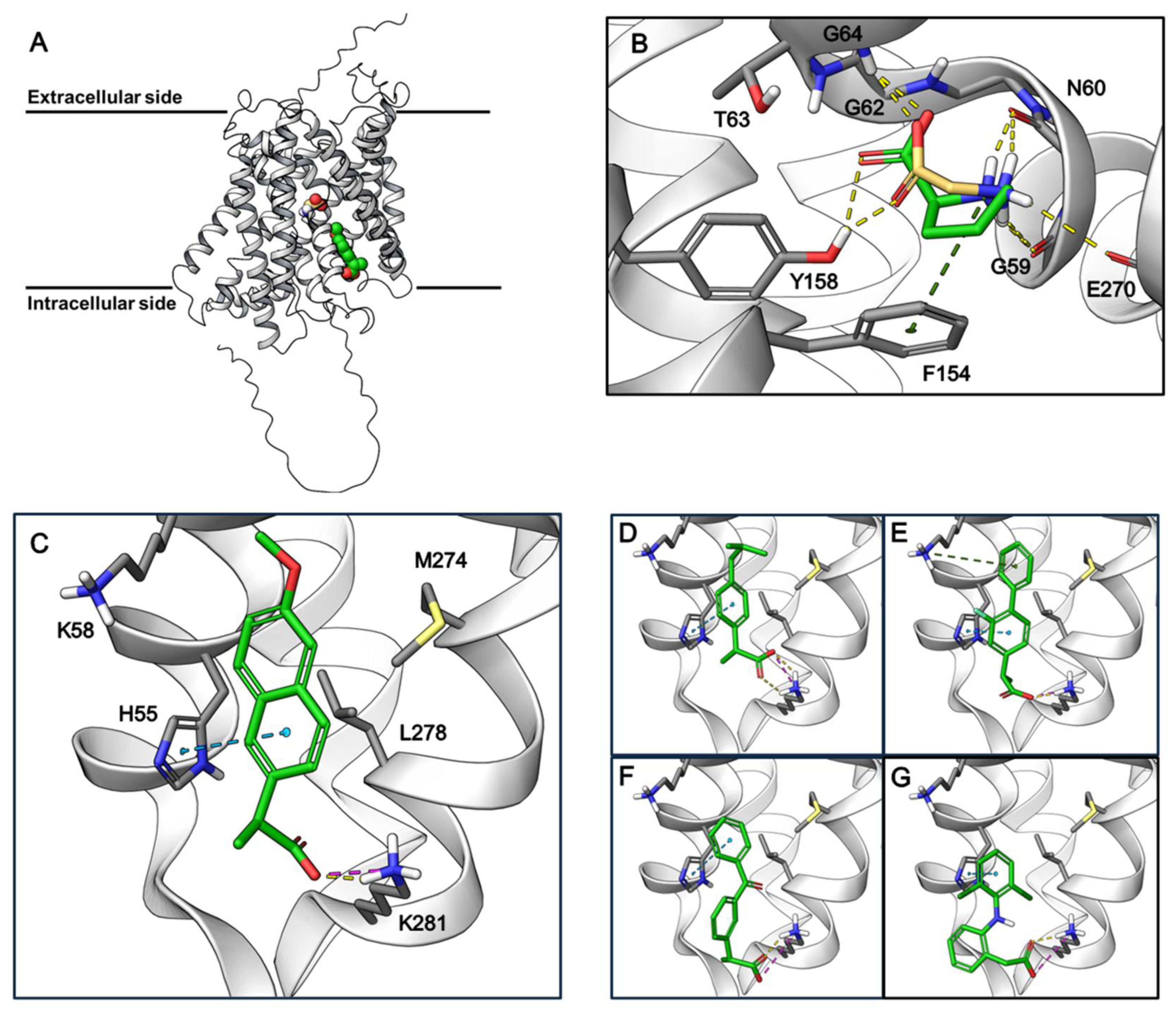
3.4. Proposed Binding Sites of Amino Acids and NSAIDs in the AlphaFold2 Model of hPAT1
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies, N.M. Clinical pharmacokinetics of ibuprofen. The first 30 years. Clin. Pharmacokinet. 1998, 34, 101–154. [Google Scholar] [CrossRef] [PubMed]
- Omkvist, D.H.; Brodin, B.; Nielsen, C.U. Ibuprofen is a non-competitive inhibitor of the peptide transporter hPEPT1 (SLC15A1): Possible interactions between hPEPT1 substrates and ibuprofen. Br. J. Pharmacol. 2010, 161, 1793–1805. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343 Pt 2, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Gopal, E.; Martin, P.M.; Itagaki, S.; Miyauchi, S.; Prasad, P.D. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008, 10, 193–199. [Google Scholar] [CrossRef]
- Itagaki, S.; Gopal, E.; Zhuang, L.; Fei, Y.J.; Miyauchi, S.; Prasad, P.D.; Ganapathy, V. Interaction of ibuprofen and other structurally related NSAIDs with the sodium-coupled monocarboxylate transporter SMCT1 (SLC5A8). Pharm. Res. 2006, 23, 1209–1216. [Google Scholar] [CrossRef]
- Brodin, B.; Nielsen, C.U.; Steffansen, B.; Frokjaer, S. Transport of peptidomimetic drugs by the intestinal Di/tri-peptide transporter, PepT1. Pharmacol. Toxicol. 2002, 90, 285–296. [Google Scholar] [CrossRef]
- Lagas, J.S.; van der Kruijssen, C.M.; van de Wetering, K.; Beijnen, J.H.; Schinkel, A.H. Transport of diclofenac by breast cancer resistance protein (ABCG2) and stimulation of multidrug resistance protein 2 (ABCC2)-mediated drug transport by diclofenac and benzbromarone. Drug Metab. Dispos. 2009, 37, 129–136. [Google Scholar] [CrossRef]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Zhang, Y.; Laracuente, M.L.; DeMarco, K.M.; Ronaldson, P.T.; Davis, T.P. P-glycoprotein modulates morphine uptake into the CNS: A role for the non-steroidal anti-inflammatory drug diclofenac. PLoS ONE 2014, 9, e88516. [Google Scholar] [CrossRef]
- Hamdi, B.A.; Amin, Z.A.; Shareef, A.M.Y.; Al-Bustany, H.A. Diclofenac sodium and dexamethasone co-therapy restores brain neuron-specific enolase (NSE), S-100 Beta and glial fibrillary acid protein (GFAP) proteins in experimental rat’s model: A possible inhibition of P-glycoprotein. Cell. Mol. Biol. 2023, 69, 100–105. [Google Scholar] [CrossRef]
- Reid, G.; Wielinga, P.; Zelcer, N.; van der Heijden, I.; Kuil, A.; de Haas, M.; Wijnholds, J.; Borst, P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2003, 100, 9244–9249. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Tirschmann, K.; Lubenow, L.E.; Nieuwenhuis, H.K.; Akkerman, J.W.; Greinacher, A.; Kroemer, H.K. The nucleotide transporter MRP4 (ABCC4) is highly expressed in human platelets and present in dense granules, indicating a role in mediator storage. Blood 2004, 104, 3603–3610. [Google Scholar] [CrossRef] [PubMed]
- El-Sheikh, A.A.; van den Heuvel, J.J.; Koenderink, J.B.; Russel, F.G. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J. Pharmacol. Exp. Ther. 2007, 320, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Berthier, J.; Benmameri, M.; Sauvage, F.L.; Fabre, G.; Chantemargue, B.; Arnion, H.; Marquet, P.; Trouillas, P.; Picard, N.; Saint-Marcoux, F. MRP4 is responsible for the efflux transport of mycophenolic acid beta-d glucuronide (MPAG) from hepatocytes to blood. Xenobiotica 2021, 51, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Mulato, A.S.; Ho, E.S.; Cihlar, T. Nonsteroidal anti-inflammatory drugs efficiently reduce the transport and cytotoxicity of adefovir mediated by the human renal organic anion transporter 1. J. Pharmacol. Exp. Ther. 2000, 295, 10–15. [Google Scholar]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J. Pharmacol. Exp. Ther. 2002, 302, 666–671. [Google Scholar] [CrossRef]
- Wood, M.; Morales, M.; Miller, E.; Braziel, S.; Giancaspro, J.; Scollan, P.; Rosario, J.; Gayapa, A.; Krmic, M.; Lee, S. Ibuprofen and the Phosphatidylcholine Bilayer: Membrane Water Permeability in the Presence and Absence of Cholesterol. Langmuir 2021, 37, 4468–4480. [Google Scholar] [CrossRef]
- Kremkow, J.; Luck, M.; Huster, D.; Muller, P.; Scheidt, H.A. Membrane Interaction of Ibuprofen with Cholesterol-Containing Lipid Membranes. Biomolecules 2020, 10, 1384. [Google Scholar] [CrossRef]
- Frolund, S.; Marquez, O.C.; Larsen, M.; Brodin, B.; Nielsen, C.U. Delta-aminolevulinic acid is a substrate for the amino acid transporter SLC36A1 (hPAT1). Br. J. Pharmacol. 2010, 159, 1339–1353. [Google Scholar] [CrossRef]
- Frolund, S.; Holm, R.; Brodin, B.; Nielsen, C.U. The proton-coupled amino acid transporter, SLC36A1 (hPAT1), transports Gly-Gly, Gly-Sar and other Gly-Gly mimetics. Br. J. Pharmacol. 2010, 161, 589–600. [Google Scholar] [CrossRef]
- Frolund, S.; Langthaler, L.; Kall, M.A.; Holm, R.; Nielsen, C.U. Intestinal drug transport via the proton-coupled amino acid transporter PAT1 (SLC36A1) is inhibited by Gly-X(aa) dipeptides. Mol. Pharm. 2012, 9, 2761–2769. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Frølund, S.; Nøhr, M.K.; Holm, R.; Brodin, B.; Nielsen, C.U. Potential involvement of the proton-coupled amino acid transporter PAT1 (SLC36A1) in the delivery of pharmaceutical agents. J. Drug Deliv. Sci. Technol. 2013, 23, 293–306. [Google Scholar] [CrossRef]
- Chen, Z.; Fei, Y.J.; Anderson, C.M.; Wake, K.A.; Miyauchi, S.; Huang, W.; Thwaites, D.T.; Ganapathy, V. Structure, function and immunolocalization of a proton-coupled amino acid transporter (hPAT1) in the human intestinal cell line Caco-2. J. Physiol. 2003, 546, 349–361. [Google Scholar] [CrossRef]
- Nielsen, C.U.; Pedersen, M.; Muller, S.; Kaestel, T.; Bjerg, M.; Ulaganathan, N.; Nielsen, S.; Carlsen, K.L.; Nohr, M.K.; Holm, R. Inhibitory Effects of 17-alpha-Ethinyl-Estradiol and 17-beta-Estradiol on Transport Via the Intestinal Proton-Coupled Amino Acid Transporter (PAT1) Investigated In Vitro and In Vivo. J. Pharm. Sci. 2021, 110, 354–364. [Google Scholar] [CrossRef]
- Schrödinger Release 2023-2; Maestro, S., LLC: New York, NY, USA, 2024.
- Colas, C. Toward a Systematic Structural and Functional Annotation of Solute Carriers Transporters-Example of the SLC6 and SLC7 Families. Front. Pharmacol. 2020, 11, 1229. [Google Scholar] [CrossRef]
- Edwards, N.; Anderson, C.M.H.; Conlon, N.J.; Watson, A.K.; Hall, R.J.; Cheek, T.R.; Embley, T.M.; Thwaites, D.T. Resculpting the binding pocket of APC superfamily LeuT-fold amino acid transporters. Cell. Mol. Life Sci. 2018, 75, 921–938. [Google Scholar] [CrossRef]
- Metzner, L.; Kottra, G.; Neubert, K.; Daniel, H.; Brandsch, M. Serotonin, L-tryptophan, and tryptamine are effective inhibitors of the amino acid transport system PAT1. FASEB J. 2005, 19, 1468–1473. [Google Scholar] [CrossRef]
- Legen, I.; Zakelj, S.; Kristl, A. Polarised transport of monocarboxylic acid type drugs across rat jejunum in vitro: The effect of mucolysis and ATP-depletion. Int. J. Pharm. 2003, 256, 161–166. [Google Scholar] [CrossRef]
- Metzner, L.; Natho, K.; Zebisch, K.; Dorn, M.; Bosse-Doenecke, E.; Ganapathy, V.; Brandsch, M. Mutational analysis of histidine residues in the human proton-coupled amino acid transporter PAT1. Biochim. Biophys. Acta 2008, 1778, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- FDA. In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. Available online: https://www.fda.gov/media/135587/download (accessed on 15 November 2024).
- Agency, E.M. Guideline on the Investigation of Drug Interactions. CPMP/EWP/560/95/Rev. 1 Corr. 2. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 29 November 2024).
- Bushra, R.; Aslam, N. An overview of clinical pharmacology of Ibuprofen. Oman Med. J. 2010, 25, 155–1661. [Google Scholar] [CrossRef] [PubMed]
- Broberg, M.; Holm, R.; Tonsberg, H.; Frolund, S.; Ewon, K.B.; Nielsen, A.; Brodin, B.; Jensen, A.; Kall, M.A.; Christensen, K.V.; et al. Function and expression of the proton-coupled amino acid transporter PAT1 along the rat gastrointestinal tract: Implications for intestinal absorption of gaboxadol. Br. J. Pharmacol. 2012, 167, 654–665. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ibuprofen | Km, app (mM) | Vmax, app (nmol cm−2 min −1) | Intrinsic CL (µL min−1 pr. cm2) |
|---|---|---|---|
| +0.0 mM | 5.4 ± 2.2 | 2.2 ± 0.4 | 0.4 |
| +0.5 mM | 3.9 ± 1.9 | 0.9± 0.2 * | 0.2 |
| +1.0 mM | 5.2 ± 3.7 | 0.9 ± 0.3 * | 0.2 |
| +2.0 mM | ND | ND | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nielsen, C.U.; Jakobsen, S.; Pedersen, M.L. Non-Steroidal Anti-Inflammatory Drugs Are Inhibitors of the Intestinal Proton-Coupled Amino Acid Transporter (PAT1): Ibuprofen and Diclofenac Are Non-Translocated Inhibitors. Pharmaceutics 2025, 17, 49. https://doi.org/10.3390/pharmaceutics17010049
Nielsen CU, Jakobsen S, Pedersen ML. Non-Steroidal Anti-Inflammatory Drugs Are Inhibitors of the Intestinal Proton-Coupled Amino Acid Transporter (PAT1): Ibuprofen and Diclofenac Are Non-Translocated Inhibitors. Pharmaceutics. 2025; 17(1):49. https://doi.org/10.3390/pharmaceutics17010049
Chicago/Turabian StyleNielsen, Carsten Uhd, Sebastian Jakobsen, and Maria L. Pedersen. 2025. "Non-Steroidal Anti-Inflammatory Drugs Are Inhibitors of the Intestinal Proton-Coupled Amino Acid Transporter (PAT1): Ibuprofen and Diclofenac Are Non-Translocated Inhibitors" Pharmaceutics 17, no. 1: 49. https://doi.org/10.3390/pharmaceutics17010049
APA StyleNielsen, C. U., Jakobsen, S., & Pedersen, M. L. (2025). Non-Steroidal Anti-Inflammatory Drugs Are Inhibitors of the Intestinal Proton-Coupled Amino Acid Transporter (PAT1): Ibuprofen and Diclofenac Are Non-Translocated Inhibitors. Pharmaceutics, 17(1), 49. https://doi.org/10.3390/pharmaceutics17010049