
Bilosomal Co-Encapsulated Tamoxifen and Propranolol for Potentiated Anti-Breast Cancer Efficacy: In Vitro and In Vivo Investigation
 ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Spectrophotometric Quantification of TAM and PRO
2.3. Preparation of Standard Niosomes and Bilosomes
2.4. Transmission Electron Microscopy
2.5. Determination of Particle Size and Zeta Potential
2.6. Determination of Entrapment Efficiency
2.7. In Vitro Drug Release
2.8. In Vivo Evaluation of Anti-Tumor Activity
2.8.1. Experimental Design
2.8.2. Tumor Volume (V) and Percentage of Tumor Growth Inhibition (% TGI) Measurement
2.8.3. Histopathological Examination of Tumor Tissues
2.9. Statistical Analysis
3. Results and Discussion
3.1. Spectrophotometric Determination of Drugs
3.2. Transmission Electron Microscopy (TEM)
3.3. Particle Size and Zeta Potential
3.4. Determination of Entrapment Efficiency
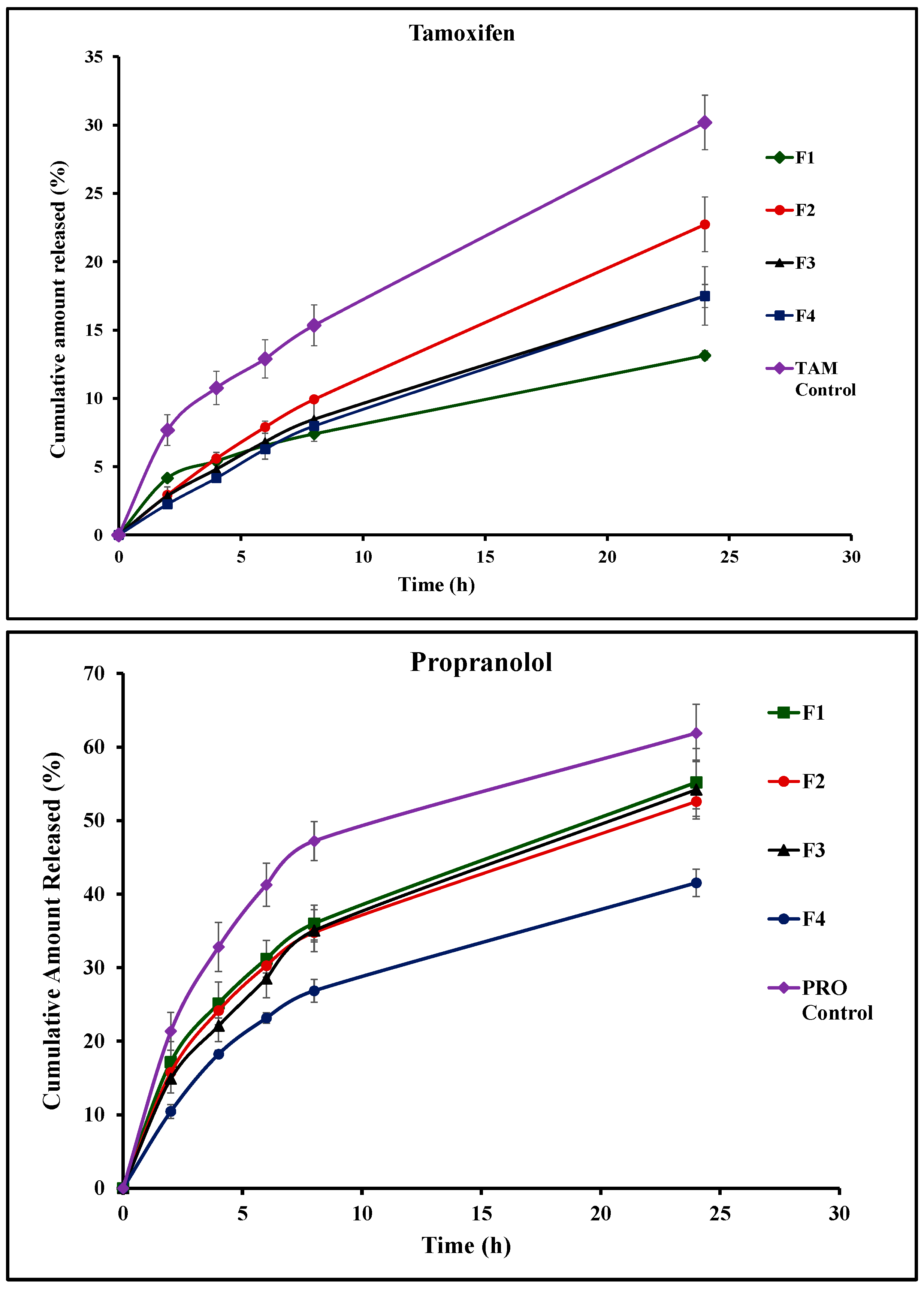
3.5. In Vitro Drug Release
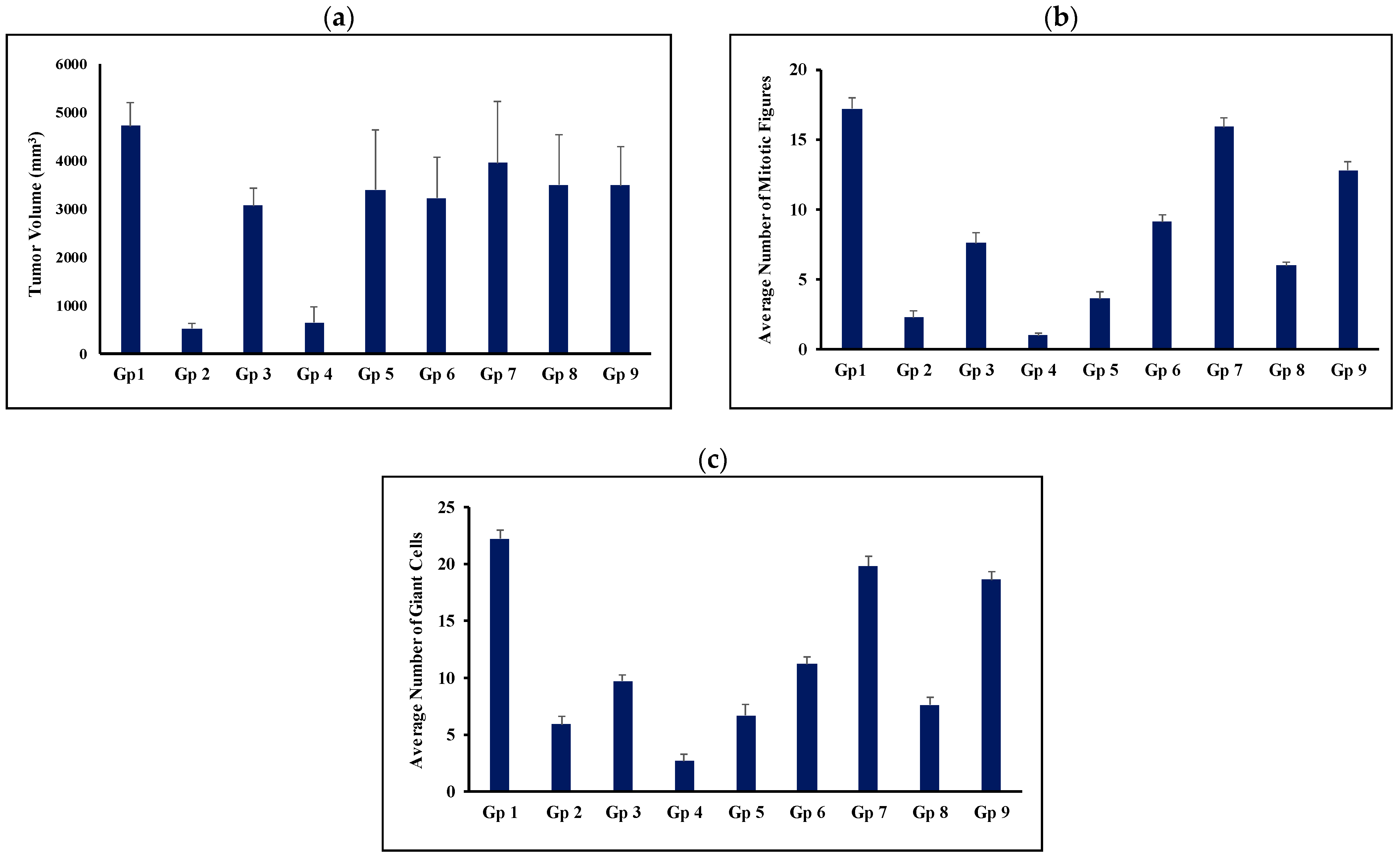
3.6. In Vivo Evaluation of Anti-Tumor Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kinnel, B.; Singh, S.K.; Oprea-Ilies, G.; Singh, R. Targeted Therapy and Mechanisms of Drug Resistance in Breast Cancer. Cancers 2023, 15, 1320. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Mailankody, S. Research and Development Spending to Bring a Single Cancer Drug to Market and Revenues After Approval. JAMA Intern. Med. 2017, 177, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-itraconazole as an anti-cancer agent. Ecancermedicalscience 2015, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Degoute, C.S. Controlled hypotension: A guide to drug choice. Drugs 2007, 67, 1053–1076. [Google Scholar] [CrossRef]
- Westfall, T.C.; Macarthur, H.; Westfall, D.P. Adrenergic agonists and antagonists. In Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13th ed.; Brunton, L.L., Hilal-Dandan, R., Knollmann, B.C., Eds.; McGraw Hill: New York, NY, USA, 2017; Available online: https://accessmedicine.mhmedical.com/Content.aspx?bookId=2189§ionId=167890123 (accessed on 10 August 2023).
- Albiñana, V.; Escribano, R.M.J.; Soler, I.; Padial, L.R.; Recio-Poveda, L.; Villar Gómez de Las Heras, K.; Botella, L.M. Repurposing propranolol as a drug for the treatment of retinal haemangioblastomas in von Hippel-Lindau disease. Orphanet J. Rare Dis. 2017, 12, 122. [Google Scholar] [CrossRef]
- González-Rodríguez, B.; Villar Gómez de Las Heras, K.; Aguirre, D.T.; Rodríguez-Padial, L.; Albiñana, V.; Recio-Poveda, L.; Cuesta, A.M.; Botella, L.M.; Jiménez-Escribano, R.M. Evaluation of the safety and effectiveness of oral propranolol in patients with von Hippel-Lindau disease and retinal hemangioblastomas: Phase III clinical trial. BMJ Open Ophthalmol. 2019, 4, e000203. [Google Scholar] [CrossRef]
- Albiñana, V.; Cuesta, A.M.; de Rojas-P, I.; Gallardo-Vara, E.; Recio-Poveda, L.; Bernabéu, C.; Botella, L.M. Review of Pharmacological Strategies with Repurposed Drugs for Hereditary Hemorrhagic Telangiectasia Related Bleeding. J. Clin. Med. 2020, 9, 1766. [Google Scholar] [CrossRef]
- Cuesta, A.M.; Gallardo-Vara, E.; Casado-Vela, J.; Recio-Poveda, L.; Botella, L.M.; Albiñana, V. The Role of Propranolol as a Repurposed Drug in Rare Vascular Diseases. Int. J. Mol. Sci. 2022, 23, 4217. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef]
- Vogelpoel, H.; Welink, J.; Amidon, G.L.; Junginger, H.E.; Midha, K.K.; Möller, H.; Olling, M.; Shah, V.; Barends, D. Biowaiver monographs for immediate release solid oral dosage forms based on biopharmaceutics classification system (BCS) literature data: Verapamil hydrochloride, propranolol hydrochloride, and atenolol. J. Pharm. Sci. 2004, 93, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Sukhatme, V.; Meheus, L.; Rooman, I.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-Propranolol as an anti-cancer agent. Ecancermedicalscience 2016, 10, 680. [Google Scholar] [CrossRef]
- O’Regan, R.M.; Jordan, V.C. The evolution of tamoxifen therapy in breast cancer: Selective oestrogen-receptor modulators and down regulators. Lancet Oncol. 2002, 3, 207–214. [Google Scholar] [CrossRef]
- Powles, T.J. Anti-oestrogenic chemoprevention of breast cancer-the need to progress. Eur. J. Cancer 2003, 39, 572–579. [Google Scholar] [CrossRef]
- Traboulsi, T.; El Ezzy, M.; Gleason, J.L.; Mader, S. Antiestrogens: Structure-activity relationships and use in breast cancer treatment. J. Mol. Endocrinol. 2017, 58, R15–R31. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef]
- Shin, S.; Choi, J.; Xiuguo, L. Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int. J. Pharm. 2006, 313, 144–149. [Google Scholar] [CrossRef]
- Sultan, A.A.; El-Gizawy, S.A.; Osman, M.A.; El Maghraby, G.M. Colloidal carriers for extended absorption window of furosemide. J. Pharm. Pharmacol. 2016, 68, 324–332. [Google Scholar] [CrossRef]
- Fayed, N.D.; Goda, A.E.; Essa, E.A.; El Maghraby, G.M. Chitosan-encapsulated niosomes for enhanced oral delivery of atorvastatin. J. Drug Deliv. Sci. Technol. 2021, 66, 102866. [Google Scholar] [CrossRef]
- Sultan, A.A.; El-Gizawy, S.A.; Osman, M.A.; El Maghraby, G.M. Niosomes for oral delivery of nateglinide: In situ-in vivo correlation. J. Liposome Res. 2018, 28, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.A.; Saad, G.A.; El Maghraby, G.M. Permeation enhancers loaded bilosomes for improved intestinal absorption and cytotoxic activity of doxorubicin. Int. J. Pharm. 2023, 630, 122427. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wei, M.; He, S.; Yuan, W.E. Advances of Non-Ionic Surfactant Vesicles (Niosomes) and Their Application in Drug Delivery. Pharmaceutics 2019, 11, 55. [Google Scholar] [CrossRef]
- Saad, G.; El Maghraby, G.M.; Sultan, A.A. Olive oil and flaxseed oil incorporating niosomes for enhanced in vivo anti-diabetic efficacy of canagliflozin. Drug Dev. Ind. Pharm. 2024, 50, 801–809. [Google Scholar] [CrossRef]
- Jiang, Z.; Fu, Y.; Shen, H. Development of Intratumoral Drug Delivery Based Strategies for Antitumor Therapy. Drug Des. Dev. Ther. 2024, 18, 2189–2202. [Google Scholar] [CrossRef]
- Saifi, O.; Rizwanullah, M.; Mir, S.R.; Amin, S. Bilosomes nanocarriers for improved oral bioavailability of acyclovir: A complete characterization through in vitro, ex-vivo and in vivo assessment. J. Drug Deliv. Sci. Technol. 2020, 57, 101634. [Google Scholar] [CrossRef]
- Khan, K.A. The concept of dissolution efficiency. J. Pharm. Pharmacol. 1975, 27, 48–49. [Google Scholar] [CrossRef]
- Moore, J.W.; Flanner, H.H. Mathematical Comparison of curves with an emphasis on in vitro dissolution profiles. Pharm. Technol. 1996, 20, 64–74. [Google Scholar]
- Khedr, N.F.; Khalil, R.M. Effect of hesperidin on mice bearing Ehrlich solid carcinoma maintained on doxorubicin. Tumor Biol. 2015, 36, 9267–9275. [Google Scholar] [CrossRef]
- Rosenberg, I.L.; Russell, C.W.; Giles, G.R. Cell viability studies on the exfoliated colonic cancer cell. Br. J. Surg. 1978, 65, 188–190. [Google Scholar] [CrossRef]
- Osman, A.E.-M.M.; Ahmed, M.M.S.; Khayyal, M.T.E.-D.; El-Merzabani, M.M. Hyperthermic potentiation of cisplatin cytotoxicity on solid Ehrlich carcinoma. Tumori 1993, 79, 268–272. [Google Scholar] [CrossRef]
- Wei, Q.; Yao, A.; Zhang, Z.; Zhang, D.; Mai, L.; Zhou, Q. Design, synthesis, and in vitro and in vivo biological studies of a 3′-deoxythymidine conjugate that potentially kills cancer cells selectively. PLoS ONE 2012, 7, e52199. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Seol, I.; Son, C. Interpretation of animal dose and human equivalent dose for drug development. J. Korean Orient. Med. 2010, 31, 1–7. [Google Scholar]
- Xing, J.; Qi, X.; Jiang, Y.; Zhu, X.; Zhang, Z.; Qin, X.; Wu, Z. Topotecan hydrochloride liposomes incorporated into thermosensitive hydrogel for sustained and efficient in situ therapy of H22 tumor in Kunming mice. Pharm. Dev. Technol. 2014, 20, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Nakase, Y.; Hagiwara, A.; Kin, S.; Fukuda, K.; Ito, T.; Takagi, T.; Fujiyama, J.; Sakakura, C.; Otsuji, E.; Yamagishi, H. Intratumoral administration of methotrexate bound to activated carbon particles: Antitumor effectiveness against human colon carcinoma xenografts and acute toxicity in mice. J. Pharmacol. Exp. Ther. 2004, 311, 382–387. [Google Scholar] [CrossRef]
- Yang, R.K.; Kalogriopoulos, N.A.; Rakhmilevich, A.L.; Ranheim, E.A.; Seo, S.; Kim, K.; Alderson, K.L.; Gan, J.; Reisfeld, R.A.; Gillies, S.D.; et al. Intratumoral hu14.18-IL-2 (IC) induces local and systemic antitumor effects that involve both activated T and NK cells as well as enhanced IC retention. J. Immunol. 2012, 189, 2656–2664. [Google Scholar] [CrossRef]
- Van De Voort, T.J.; Felder, M.A.; Yang, R.K.; Sondel, P.M.; Rakhmilevich, A.L. Intratumoral delivery of low doses of anti-CD40 mAb combined with monophosphoryl lipid a induces local and systemic antitumor effects in immunocompetent and T cell-deficient mice. J. Immunother. 2013, 36, 29–40. [Google Scholar] [CrossRef]
- Sancéau, J.; Poupon, M.F.; Delattre, O.; Sastre-Garau, X.; Wietzerbin, J. Strong inhibition of Ewing tumor xenograft growth by combination of human interferon-alpha or interferon-beta with ifosfamide. Oncogene 2002, 21, 7700–7709. [Google Scholar] [CrossRef]
- Goda, A.E.; Elsisi, A.E.; Sokkar, S.S.; Abdelrazik, N.M. Enhanced in vivo targeting of estrogen receptor alpha signaling in murine mammary adenocarcinoma by nilotinib/rosuvastatin novel combination. Toxicol. Appl. Pharmacol. 2020, 404, 115185. [Google Scholar] [CrossRef] [PubMed]
- Souza, T.; Ciminelli, V.; Mohallem, N. A comparison of TEM and DLS methods to characterize size distribution of ceramic nanoparticles. J. Phys. Conf. Ser. 2016, 733, 012039. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Teeranachaideekul, V.; Supaperm, T. Effect of charged and non-ionic membrane additives on physicochemical properties and stability of niosomes. AAPS PharmSciTech 2008, 9, 851–859. [Google Scholar] [CrossRef] [PubMed]
- El Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. Drug interaction and location in liposomes: Correlation with polar surface areas. Int. J. Pharm. 2005, 292, 179–185. [Google Scholar] [CrossRef] [PubMed]
- El Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. Skin delivery of oestradiol from deformable and traditional liposomes: Mechanistic studies. J. Pharm. Pharmacol. 1999, 51, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- El Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. Skin delivery of 5-fluorouracil from ultradeformableand standard liposomes in-vitro. J. Pharm. Pharmacol. 2001, 53, 1069–1077. [Google Scholar] [CrossRef]
- Essa, E.A. Effect of formulation and processing variables on the particle size of sorbitan monopalmitate niosomes. Asian J. Pharm. 2010, 4, 227–233. [Google Scholar] [CrossRef]
- Moghtaderi, M.; Sedaghatnia, K.; Bourbour, M.; Fatemizadeh, M.; Salehi Moghaddam, Z.; Hejabi, F.; Heidari, F.; Quazi, S.; Farasati Far, B. Niosomes: A novel targeted drug delivery system for cancer. Med. Oncol. 2022, 39, 240. [Google Scholar] [CrossRef]
- El Maghraby, G.M.; Ahmed, A.A.; Osman, M.A. Penetration enhancers in proniosomes as a new strategy for enhanced transdermal drug delivery. Saudi Pharm. J. 2015, 23, 67–74. [Google Scholar] [CrossRef]
- Zaid Alkilani, A.; Hamed, R.; Abdo, H.; Swellmeen, L.; Basheer, H.A.; Wahdan, W.; Abu Kwiak, A.D. Formulation and Evaluation of Azithromycin-Loaded Niosomal Gel: Optimization, In Vitro Studies, Rheological Characterization, and Cytotoxicity Study. ACS Omega 2022, 7, 39782–39793. [Google Scholar] [CrossRef]
- Binsuwaidan, R.; Sultan, A.A.; Negm, W.A.; Attallah, N.G.M.; Alqahtani, M.J.; Hussein, I.A.; Shaldam, M.A.; El-Sherbeni, S.A.; Elekhnawy, E. Bilosomes as Nanoplatform for Oral Delivery and Modulated In Vivo Antimicrobial Activity of Lycopene. Pharmaceuticals 2022, 15, 1043. [Google Scholar] [CrossRef]
- SreeHarsha, N.; Hiremath, J.G.; Chilukuri, S.; Aitha, R.K.; Al-Dhubiab, B.E.; Venugopala, K.N.; Alzahrani, A.M.; Meravanige, G. An Approach to Enhance Dissolution Rate of Tamoxifen Citrate. BioMed Res. Int. 2019, 2019, 2161348. [Google Scholar] [CrossRef]
- Bolourchian, N.; Dadashzadeh, S. pH-independent release of propranolol hydrochloride from HPMC-based matrices using organic acids. DARU J. Pharm. Sci. 2008, 16, 136–142. [Google Scholar]
- Phillips, D.J.; Pygall, S.R.; Cooper, V.B.; Mann, J.C. Overcoming sink limitations in dissolution testing: A review of traditional methods and the potential utility of biphasic systems. J. Pharm. Pharmacol. 2012, 64, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- El Nashar, N.F.; Sultan, A.A.; El Maghraby, G.M. Self-dispersing self-assembling systems for controlled oral delivery of gliclazide. J. Drug Deliv. Sci. Technol. 2021, 66, 102742. [Google Scholar] [CrossRef]
- Frajacomo, F.T.T.; Padilha, C.S.; Marinello, P.C.; Guarnier, F.A.; Cecchini, R.; Duarte, J.A.R.; Deminice, R. Solid Ehrlich carcinoma reproduces functional and biological characteristics of cancer cachexia. Life Sci. 2016, 162, 47–53. [Google Scholar] [CrossRef]
- Kaurav, H.; Tripathi, M.; Kaur, S.D.; Bansal, A.; Kapoor, D.N.; Sheth, S. Emerging Trends in Bilosomes as Therapeutic Drug Delivery Systems. Pharmaceutics 2024, 16, 697. [Google Scholar] [CrossRef]
- Keshgegian, A.A.; Cnaan, A. Proliferation markers in breast carcinoma. Mitotic figure count, S-phase fraction, proliferating cell nuclear antigen, Ki-67 and MIB-1. Am. J. Clin. Pathol. 1995, 104, 42–49. [Google Scholar] [CrossRef]
- Cree, I.A.; Tan, P.H.; Travis, W.D.; Wesseling, P.; Yagi, Y.; White, V.A.; Lokuhetty, D.; Scolyer, R.A. Counting mitoses: SI (ze) matters! Mod. Pathol. 2021, 34, 1651–1657. [Google Scholar] [CrossRef]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef]
- Powe, D.G.; Voss, M.J.; Zänker, K.S.; Habashy, H.O.; Green, A.R.; Ellis, I.O.; Entschladen, F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 2010, 1, 628–638. [Google Scholar] [CrossRef]
- Barron, T.I.; Connolly, R.M.; Sharp, L.; Bennett, K.; Visvanathan, K. Beta blockers and breast cancer mortality: A population- based study. J. Clin. Oncol. 2011, 29, 2635–2644. [Google Scholar] [CrossRef]
- Campbell, J.P.; Karolak, M.R.; Ma, Y.; Perrien, D.S.; Masood-Campbell, S.K.; Penner, N.L.; Munoz, S.A.; Zijlstra, A.; Yang, X.; Sterling, J.A.; et al. Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol. 2012, 10, e1001363. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.A.; El-Gizawy, S.A.; Osman, M.A.; El Maghraby, G.M. Peceosomes for oral delivery of glibenclamide: In vitro in situ correlation. J. Drug Deliv. Sci. Technol. 2017, 41, 303–309. [Google Scholar] [CrossRef]
- Faheim, S.H.; El Maghraby, G.M.; Sultan, A.A. Vesicular Carriers for Improved Oral Anticoagulation Competence of Rivaroxaban: In Vitro and In Vivo Investigation. AAPS PharmSciTech 2024, 25, 264. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Teiama, M.; Magdy, B.; Sakran, W. Development of a Novel Bilosomal System for Improved Oral Bioavailability of Sertraline Hydrochloride: Formulation Design, In Vitro Characterization, and Ex Vivo and In Vivo Studies. AAPS PharmSciTech 2022, 23, 188. [Google Scholar] [CrossRef] [PubMed]
- Arzani, G.; Haeri, A.; Daeihamed, M.; Bakhtiari-Kaboutaraki, H.; Dadashzadeh, S. Niosomal carriers enhance oral bioavailability of carvedilol: Effects of bile salt-enriched vesicles and carrier surface charge. Int. J. Nanomed. 2015, 10, 4797–4813. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Span 60 (mg) | Cholesterol (mg) | Bile Salts (mg) | Ethanol (mL) | |
|---|---|---|---|---|
| F1 | 600 | 100 | - | 1 |
| F2 | 600 | 100 | 100 | 1 |
| F3 | 600 | 100 | 150 | 1 |
| F4 | 600 | 100 | 200 | 1 |
| Group (Gp) | Drug/s | Formulation | Route of Administration | Dose Frequency | TGI (%) | Necrosis Score |
|---|---|---|---|---|---|---|
| Gp 1 | - | Vehicle | Oral | Daily | - | - |
| Gp 2 | TAM | Bilosomes | Oral | Daily | 91.6 | Grade 3 |
| Gp 3 | TAM | Suspension | Oral | Daily | 12.7 | Grade 2 |
| Gp 4 | TAM + PRO | Bilosomes | Oral | Daily | 86.9 | Grade 4 |
| Gp 5 | TAM + PRO | Suspension | Oral | Daily | 18.4 | Grade 3 |
| Gp 6 | TAM | Bilosomes | Intratumor | Once/week | 32.4 | Grade 2 |
| Gp 7 | TAM | Suspension | Intratumor | Once/week | 3.0 | Grade 1 |
| Gp 8 | TAM + PRO | Bilosomes | Intratumor | Once/week | 20.8 | Grade 2 |
| Gp 9 | TAM + PRO | Suspension | Intratumor | Once/week | 19.0 | Grade 1 |
| Formulation | EE (%) | Particle Size (nm) | PDI | ZP (mv) | RE (%) | |||
|---|---|---|---|---|---|---|---|---|
| TAM | PRO | TEM | PCS | TAM | PRO | |||
| F1 | 87.5 ± 2.4 | 45.3 ± 4.4 | 76.8 ± 28.5 | 103.9 ± 45.5 | 0.36 | −51.3 ± 7.4 | 8.4 ±0.1 | 38.0 ± 2.8 |
| F2 | 97.8 ± 0.2 | 43.6 ± 1.6 | 110.3 ± 36.5 | 145.9 ± 55.1 | 0.34 | −48.4 ± 3.0 | 11.7 ± 1.8 | 36.0 ± 3.2 |
| F3 | 95.3 ± 0.9 | 46.8 ± 1.8 | 97.8 ± 32.0 | 141.6 ± 58.8 | 0.32 | −51.7 ± 2.9 | 9.4 ± 1.5 | 36.7 ± 3.3 |
| F4 | 93.5 ± 0.4 | 31.0 ± 0.7 | 150.9 ± 33.2 | 181.7 ± 56.8 | 0.33 | −54.9 ± 4.0 | 9.8 ± 0.5 | 28.0 ± 1.3 |
| Formulation | Release Kinetics (R2 Value) | |||||
|---|---|---|---|---|---|---|
| Tamoxifen | Propranolol | |||||
| Zero | First | Higuchi | Zero | First | Higuchi | |
| F1 | 0.988 | 0.923 | 0.997 | 0.723 | 0.646 | 0.838 |
| F2 | 0.992 | 0.878 | 0.995 | 0.897 | 0.769 | 0.965 |
| F3 | 0.984 | 0.838 | 0.999 | 0.822 | 0.700 | 0.912 |
| F4 | 0.986 | 0.825 | 0.997 | 0.866 | 0.742 | 0.946 |
| Tumor Volume (mm3) | |||||||||
| Gp 1 | Gp 2 | Gp 3 | Gp 4 | Gp 5 | Gp 6 | Gp 7 | Gp 8 | Gp 9 | |
| Gp 1 | - | 0.000 | 0.005 | 0.000 | 0.031 | 0.124 | 0.699 | 0.269 | 0.264 |
| Gp 2 | 0.000 | - | 0.000 | 0.998 | 0.000 | 0.001 | 0.000 | 0.000 | 0.000 |
| Gp 3 | 0.005 | 0.000 | - | 0.000 | 0.919 | 1.000 | 0.729 | 0.993 | 0.993 |
| Gp 4 | 0.000 | 0.998 | 0.000 | - | 0.000 | 0.001 | 0.000 | 0.000 | 0.000 |
| Gp 5 | 0.031 | 0.000 | 0.919 | 0.000 | - | 1.000 | 0.969 | 1.000 | 1.000 |
| Gp 6 | 0.124 | 0.001 | 1.000 | 0.001 | 1.000 | - | 0.731 | 0.991 | 0.991 |
| Gp 7 | 0.699 | 0.000 | 0.729 | 0.000 | 0.969 | 0.731 | - | 0.933 | 0.930 |
| Gp 8 | 0.269 | 0.000 | 0.993 | 0.000 | 1.000 | 0.991 | 0.933 | - | 1.00 |
| Gp 9 | 0.264 | 0.000 | 0.993 | 0.000 | 1.000 | 0.991 | 0.930 | 1.00 | - |
| Mitotic Figures | |||||||||
| Gp 1 | Gp 2 | Gp 3 | Gp 4 | Gp 5 | Gp 6 | Gp 7 | Gp 8 | Gp 9 | |
| Gp 1 | - | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.013 | 0.000 | 0.000 |
| Gp 2 | 0.000 | - | 0.000 | 0.015 | 0.010 | 0.000 | 0.000 | 0.000 | 0.000 |
| Gp 3 | 0.000 | 0.000 | - | 0.000 | 0.000 | 0.001 | 0.000 | 0.002 | 0.000 |
| Gp 4 | 0.000 | 0.015 | 0.000 | - | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Gp 5 | 0.000 | 0.010 | 0.000 | 0.000 | - | 0.000 | 0.000 | 0.000 | 0.000 |
| Gp 6 | 0.000 | 0.000 | 0.001 | 0.000 | 0.000 | - | 0.000 | 0.000 | 0.000 |
| Gp 7 | 0.013 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | - | 0.000 | 0.000 |
| Gp 8 | 0.000 | 0.000 | 0.002 | 0.000 | 0.000 | 0.000 | 0.000 | - | 0.000 |
| Gp 9 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | - |
| Giant Cell Count | |||||||||
| Gp 1 | Gp 2 | Gp 3 | Gp 4 | Gp 5 | Gp 6 | Gp 7 | Gp 8 | Gp 9 | |
| Gp 1 | - | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Gp 2 | 0.000 | - | 0.000 | 0.000 | 0.335 | 0.000 | 0.000 | 0.000 | 0.015 |
| Gp 3 | 0.000 | 0.000 | - | 0.000 | 0.000 | 0.040 | 0.000 | 0.001 | 0.000 |
| Gp 4 | 0.000 | 0.000 | 0.000 | - | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Gp 5 | 0.000 | 0.335 | 0.000 | 0.000 | - | 0.000 | 0.000 | 0.611 | 0.000 |
| Gp 6 | 0.000 | 0.000 | 0.040 | 0.000 | 0.000 | - | 0.000 | 0.000 | 0.000 |
| Gp 7 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | - | 0.000 | 0.131 |
| Gp 8 | 0.000 | 0.000 | 0.001 | 0.000 | 0.611 | 0.000 | 0.000 | - | 0.000 |
| Gp 9 | 0.000 | 0.015 | 0.000 | 0.000 | 0.000 | 0.000 | 0.131 | 0.000 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elebyary, T.T.; Sultan, A.A.; Abu-Risha, S.E.; El Maghraby, G.M.; Amin, M. Bilosomal Co-Encapsulated Tamoxifen and Propranolol for Potentiated Anti-Breast Cancer Efficacy: In Vitro and In Vivo Investigation. Pharmaceutics 2025, 17, 123. https://doi.org/10.3390/pharmaceutics17010123
Elebyary TT, Sultan AA, Abu-Risha SE, El Maghraby GM, Amin M. Bilosomal Co-Encapsulated Tamoxifen and Propranolol for Potentiated Anti-Breast Cancer Efficacy: In Vitro and In Vivo Investigation. Pharmaceutics. 2025; 17(1):123. https://doi.org/10.3390/pharmaceutics17010123
Chicago/Turabian StyleElebyary, Toka T., Amal A. Sultan, Sally E. Abu-Risha, Gamal M. El Maghraby, and Manna Amin. 2025. "Bilosomal Co-Encapsulated Tamoxifen and Propranolol for Potentiated Anti-Breast Cancer Efficacy: In Vitro and In Vivo Investigation" Pharmaceutics 17, no. 1: 123. https://doi.org/10.3390/pharmaceutics17010123
APA StyleElebyary, T. T., Sultan, A. A., Abu-Risha, S. E., El Maghraby, G. M., & Amin, M. (2025). Bilosomal Co-Encapsulated Tamoxifen and Propranolol for Potentiated Anti-Breast Cancer Efficacy: In Vitro and In Vivo Investigation. Pharmaceutics, 17(1), 123. https://doi.org/10.3390/pharmaceutics17010123