
Applying Physiologically Based Pharmacokinetic Modeling to Interpret Carbamazepine’s Nonlinear Pharmacokinetics and Its Induction Potential on Cytochrome P450 3A4 and Cytochrome P450 2C9 Enzymes

 , , ,
, , , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Software
2.2. Clinical Data
2.3. Model Development and Verification
2.4. Model Evaluation
2.5. Parameter Local Sensitivity Analysis
2.6. DDI Application and Evaluation
2.7. CBZ Nonlinear PK Exploration
3. Results
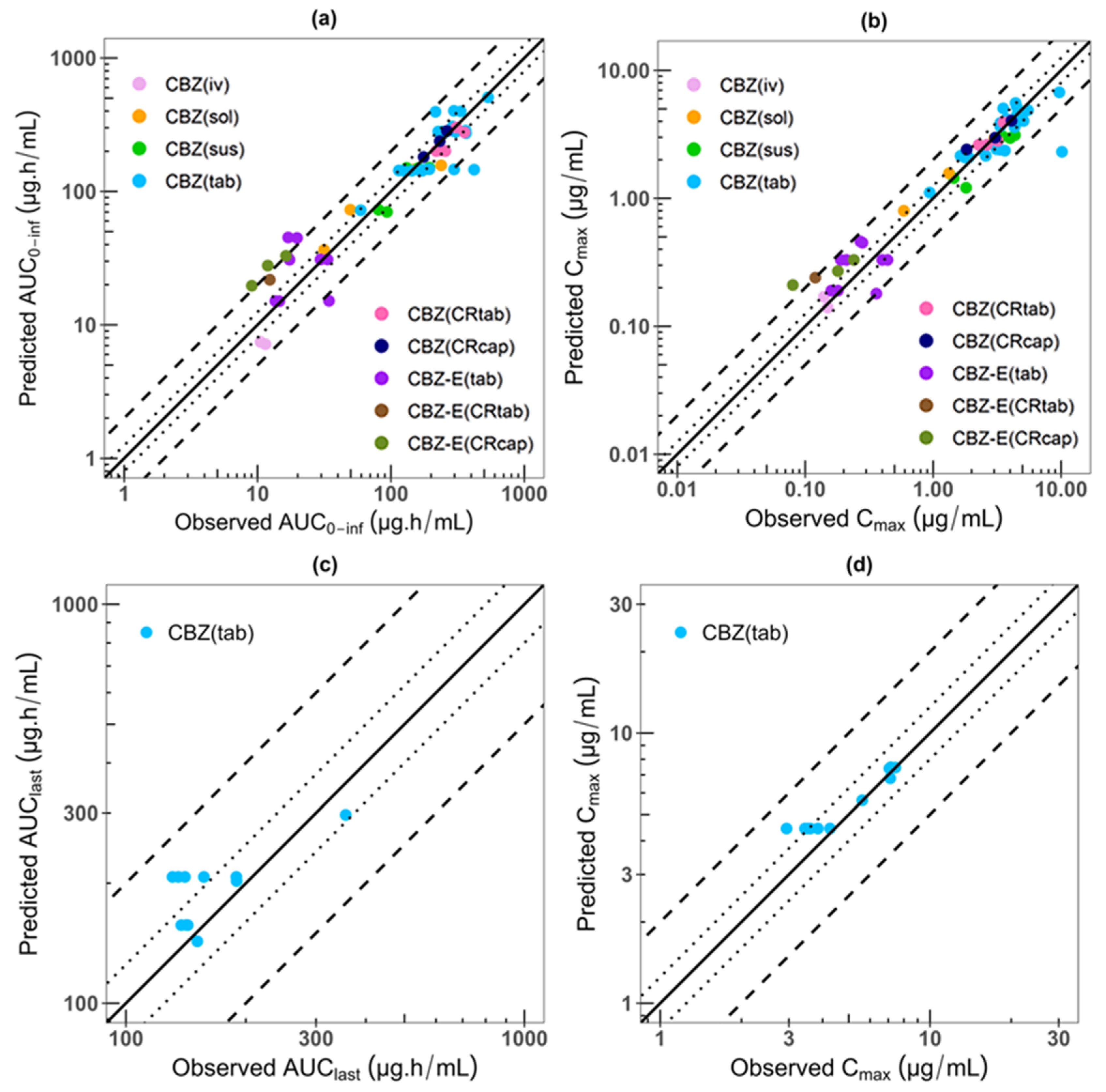
3.1. CBZ-E PBPK Model Building and Performance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Model Value | Literature Value | |
|---|---|---|---|
| Carbamazepine drug-dependent parameters | |||
| MW (g/mol) | 236.27 | - | |
| logP | 2 | 1.51 [47], 2.45 [48], 2.19 [49], 2.29 [50], 2.93 [51] | |
| fup (%) | 22.5 | 22.5–25.8 [52], 26–29.8 [8] | |
| Blood: plasma concentration ratio (Rbp) | 0.9 | 1.06 ± 0.21 [53], 0.9 ± 0.11 [54], 1.36 ± 0.10 [54] | |
| pKa | 10.86 | 10.86 (ADMET Predictor v.10.3.0.0), 11.83 [55], 14 [47] | |
| CYP2C8 → CBZ-E | Km (µM) | 757 | 757 [56] |
| Vmax (nmol/min/nmol CYP) | 0.669 | 0.669 [56] | |
| CYP3A4 → CBZ-E | Km (µM) | 248 | 119 [56], 248 [10], 442 [13], 630 [57] |
| Vmax (pmol/min/pmol CYP) | 0.75 1 | 1.17 [56], 4.87 [10], 1.37 [13], 5.3 [57] | |
| CYP3A5 → CBZ-E | Km (µM) | 2300 | 2300 [10], 338 [57] |
| Vmax (pmol/min/pmol CYP) | 10 | 10 [10], 5.98 [57] | |
| CYP2B6 → OH-CBZ | Km (µM) | 420 | 420 [11] |
| Vmax (pmol/min/pmol CYP) | 0.429 | 0.429 [11] | |
| CYP3A4 → OH-CBZ | Km (µM) | 282 | 282 [11] |
| Vmax (pmol/min/pmol CYP) | 0.164 | 0.164 [11] | |
| UGT2B7 → CBZ-glu | Km (µM) | 214 | 214 [15] |
| Vmax (pmol/min/mg) | 0.79 | 0.79 [15] | |
| CLint,liver,unbound (L/h) | 3.316 2 | - | |
| CLrenal,filt (L/h) | 0.0084 | 0.0084 [16] | |
| Peff (cm/s × 10−4) | 4.3 | 4.3 ± 2.7 [58,59] | |
| Aqueous solubility (mg/mL, pH) | 0.127 (6.5) | 0.12(6.8) [37], 0.127(6.5) [60], 0.214(1) [61], 0.26(1) [39] | |
| Solubility (mg/mL, SGF at pH = 1.2 at 0 mM) | 0.236 | 0.236 [60] | |
| Solubility (mg/mL, FaSSIF at pH = 6.8 at 3 mM) | 0.283 | 0.132 [60], 0.234 [62], 0.24 [3], 0.27 [63], 0.283 [60], 0.31 [63] | |
| Solubility (mg/mL, FeSSIF at pH = 6.8 at 15 mM) | 0.52 | 0.343 [62], 0.47 [63], 0.52 [63] | |
| Diffusion Coefficient (cm2/s × 105) | 0.86 | ADMET Predictor v.10.3.0.0 | |
| CBZ formulation parameters | |||
| Solution and suspension | |||
| Particle density (g/mL) | 1.2 | GastroPlus® default value | |
| Mean Particle Radius (µm) | 25 | GastroPlus® default value | |
| Particle radius standard deviation | 0 | GastroPlus® default value | |
| Particle radius bin# | 1 | GastroPlus® default value | |
| IR tablet | |||
| Particle density (g/mL) | 1.5 | 1.5 [37] | |
| Mean Particle Radius (µm) | 60 | 75 [37], 1–40 [38], 62.5–100 [39], 7.5–168 [40] | |
| Particle Radius standard deviation | 20 | 20 [37] | |
| Particle radius bin# | 5 | 5 [37] | |
| CR tablet 3 | |||
| T (time lag) (h) | 0.5 | Optimized value | |
| Max (total release) (%) | 95 | Optimized value | |
| A (time scale) (hrsb) | 3 | Optimized value | |
| B (shape) | 0.45 (fast), 1 (fed) | Optimized value | |
| CR capsule 3 | |||
| T (time lag) (h) | 0.7 | Optimized value | |
| Max (total release) (%) | 100 | Optimized value | |
| A (time scale) (hrsb) | 4.7 | Optimized value | |
| b (shape) | 0.8 (fast), 1.4 (fed) | Optimized value | |
| Carbamazepine-10,11-epoxide drug-dependent parameters | |||
| MW (g/mol) | 252.27 | - | |
| logP | 1.4 | 1.58 [64], 1.97 [64] | |
| Fup (%) | 51.8 | 46.8–51.8 [7] | |
| Blood: plasma concentration ratio (Rbp) | 1.53 | 1.53 ± 0.45 [53], 1.27–1.80 [54] | |
| pKa | 11.03 | ADMET Predictor v.10.3.0.0 | |
| CLhepatic (L/h) | 4.86 4 | - | |
| CLrenal,filt (L/h) | 0.14 | 0.14 [16] | |
| Peff (cm/s × 10−4) | 50 | 50 [43] | |
| Solubility in water (assume pH = 7, mg/mL) | 1.34 | 1.34 [64] | |
| Diffusion Coefficient (cm2/s × 105) | 0.87 | ADMET Predictor v.10.3.0.0 | |
| CBZ-E formulation parameters | |||
| Solution and suspension | |||
| Particle density (g/mL) | 1.2 | GastroPlus® default value | |
| Mean Particle Radius (µm) | 25 | GastroPlus® default value | |
| Particle radius standard deviation | 0 | GastroPlus® default value | |
| Particle radius bin# | 1 | GastroPlus® default value | |
| Enteric-coated tablet 3 | |||
| T (time lag) (h) | 2 | Optimized value | |
| Max (total release) (%) | 84 | Optimized value | |
| A (time scale) (hrsb) | 1 | Optimized value | |
| b (shape) | 0.75 (fast) | Optimized value | |
3.2. CBZ P-M PBPK Model Building and Performance
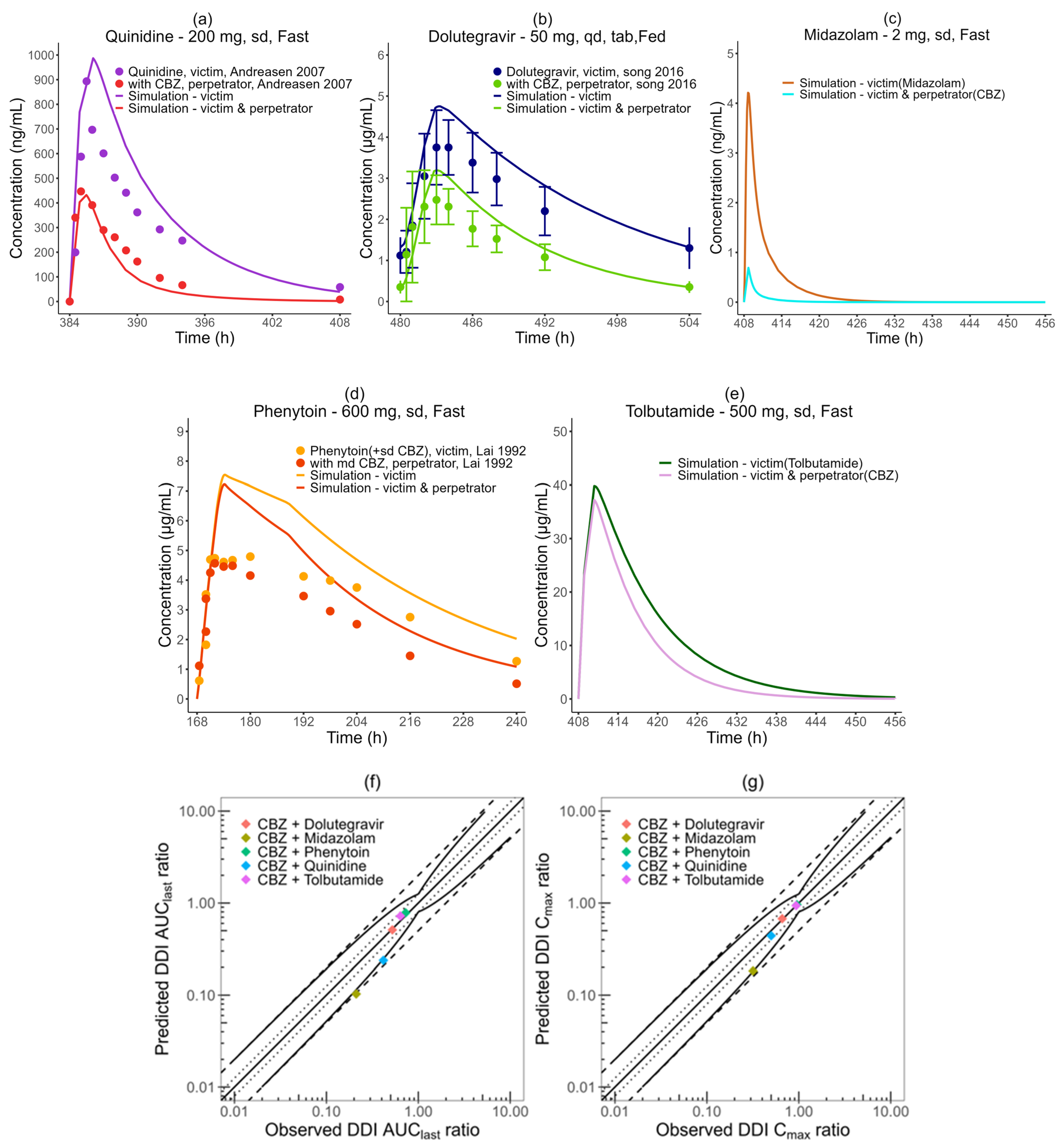
3.3. Model Application in DDI
| Enzyme | Parameters | Model Value | Literature Value |
|---|---|---|---|
| CYP3A4 | Emax | 10(Fit) | 4.57–15.73 [66], 15.6 [67], 1.9–14 [68], 6.3–31 [69], 4.7–9.7 [70], 4.1–10.7 [70], 2.3–58 [71], 2.3–40 [72], 9.3–21 [73]; 5.3–11 [74]; 3.48–14.5 [75]; 55.8/60 [76]; 3.75–11.9 [77] |
| EC50, in vitro,T (µM) | 22(Fix) | 14.37–27.70 [66], 58.7/59.1 [67], 4.3–27 [68], 40/42 [69], 13.1–27.2 [70], 10.5–16.2 [70], 12–59 [71], 16 ± 11 [42]; 29–98 [73]; 10.2/34.3 [76]; 21–28 [74]; 47.5 to 80.7 [77] | |
| CYP3A5 | Emax | 2.95 | 2.95 [42], 3.5 [72], 1.43 [78] |
| EC50, in vitro,T (µM) | 142 | 142 ± 51 [42] | |
| CYP2C8 | Emax | 3.49 | 3.49 [42]; 3.92 ± 1.34 [79] |
| EC50, in vitro,T (µM) | 22(Fix) | 22 ± 18 [42], 26.62 [79] | |
| CYP2B6 | Emax | 10.14 | 11.5–28 (mean 18.2) [80], 3.7–4.4 [81], 3.08–29.1 [75], 10.14 [42] |
| EC50, in vitro,T (µM) | 26 | 9.4–51 (mean 26) [80], 22 ± 9.7 [42] | |
| CYP2C9 | Emax | 1.83 | 1.83 [42]; 2.32 ± 0.33 [79]; 1.0–1.5 [82] |
| EC50, in vitro,T (µM) | 22(Fix) | 30 [42] |
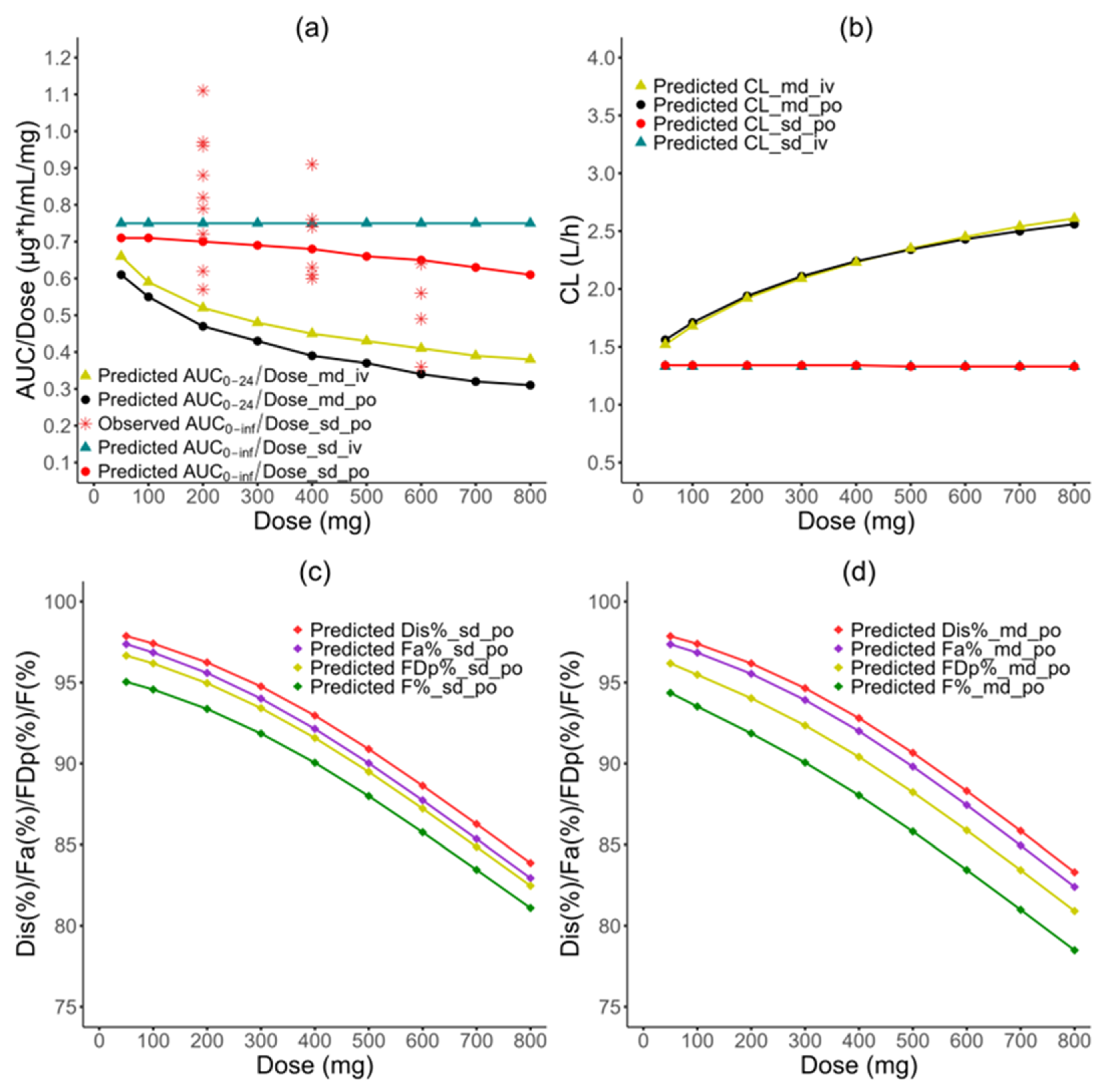
3.4. Nonlinearity PK Investigation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goldenberg, M.M. Overview of Drugs Used for Epilepsy and Seizures: Etiology, Diagnosis, and Treatment. Pharm. Ther. 2010, 35, 392–415. [Google Scholar]
- Albani, F.; Riva, R.; Baruzzi, A. Carbamazepine Clinical Pharmacology: A Review. Pharmacopsychiatry 1995, 28, 235–244. [Google Scholar] [CrossRef]
- García, M.A.; Cristofoletti, R.; Abrahamsson, B.; Groot, D.W.; Parr, A.; Polli, J.E.; Mehta, M.; Shah, V.P.; Tomakazu, T.; Dressman, J.B.; et al. Biowaiver Monograph for Immediate-Release Solid Oral Dosage Forms: Carbamazepine. J. Pharm. Sci. 2021, 110, 1935–1947. [Google Scholar] [CrossRef] [PubMed]
- Novartis Tegretol—Accessdata.Fda.Gov. Available online: https://Www.Accessdata.Fda.Gov/Drugsatfda_docs/Label/2015/016608s097,018281s045,018927s038,020234s026lbl.Pdf (accessed on 23 May 2024).
- McLean, A.; Browne, S.; Zhang, Y.; Slaughter, E.; Halstenson, C.; Couch, R. The Influence of Food on the Bioavailability of a Twice-Daily Controlled Release Carbamazepine Formulation. J. Clin. Pharmacol. 2001, 41, 183–186. [Google Scholar] [CrossRef]
- Levy, R.H.; Pitlick, W.H.; Troupin, A.S.; Green, J.R.; Neal, J.M. Pharmacokinetics of Carbamazepine in Normal Man. Clin. Pharmacol. Ther. 1975, 17, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Morselli, P.L.; Gerna, M.; de Maio, D.; Zanda, G.; Viani, F.; Garattini, S. Pharmacokinetic Studies on Carbamazepine in Volunteers and in Epileptic Patients. In Clinical Pharmacology of Anti-Epileptic Drugs; Springer: Berlin/Heidelberg, Germany, 1975; pp. 166–180. [Google Scholar]
- Rawlins, M.D.; Collste, P.; Bertilsson, L.; Palmér, L. Distribution and Elimination Kinetics of Carbamazepine in Man. Eur. J. Clin. Pharmacol. 1975, 8, 91–96. [Google Scholar] [CrossRef]
- Pynnönen, S. The Pharmacokinetics of Carbamazepine in Plasma and Saliva of Man. Acta Pharmacol. Toxicol. 1977, 41, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lin, Y.S.; McConn, D.J.; Calamia, J.C.; Totah, R.A.; Isoherranen, N.; Glodowski, M.; Thummel, K.E. Evidence of Significant Contribution from CYP3A5 to Hepatic Drug Metabolism. Drug Metab. Dispos. 2004, 32, 1434–1445. [Google Scholar] [CrossRef]
- Pearce, R.E.; Vakkalagadda, G.R.; Leeder, J.S. Pathways of Carbamazepine Bioactivation in Vitro I. Characterization of Human Cytochromes P450 Responsible for the Formation of 2- and 3-Hydroxylated Metabolites. Drug Metab. Dispos. 2002, 30, 1170–1179. [Google Scholar] [CrossRef]
- Pearce, R.E.; Uetrecht, J.P.; Leeder, J.S. Pathways of Carbamazepine Bioactivation in Vitro: II. The Role of Human Cytochrome P450 Enzymes in the Formation of 2-Hydroxyiminostilbene. Drug Metab. Dispos. 2005, 33, 1819–1826. [Google Scholar] [CrossRef]
- Kerr, B.M.; Thummel, K.E.; Wurden, C.J.; Klein, S.M.; Kroetz, D.L.; Gonzalez, F.J.; Levy, R. Human Liver Carbamazepine Metabolism. Biochem. Pharmacol. 1994, 47, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Puranik, Y.G.; Birnbaum, A.K.; Marino, S.E.; Ahmed, G.; Cloyd, J.C.; Remmel, R.P.; Leppik, I.E.; Lamba, J.K. Association of Carbamazepine Major Metabolism and Transport Pathway Gene Polymorphisms and Pharmacokinetics in Patients with Epilepsy. Pharmacogenomics 2013, 14, 35–45. [Google Scholar] [CrossRef]
- Staines, A.G.; Coughtrie, M.W.H.; Burchell, B. N-Glucuronidation of Carbamazepine in Human Tissues Is Mediated by UGT2B7. J. Pharmacol. Exp. Ther. 2004, 311, 1131–1137. [Google Scholar] [CrossRef]
- Kim, K.-A.; Oh, S.O.; Park, P.-W.; Park, J.-Y. Effect of Probenecid on the Pharmacokinetics of Carbamazepine in Healthy Subjects. Eur. J. Clin. Pharmacol. 2005, 61, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, P.N.; Berry, D.J.; Bourgeois, B.F.D.; Cloyd, J.C.; Glauser, T.A.; Johannessen, S.I.; Leppik, I.E.; Tomson, T.; Perucca, E. Antiepileptic Drugs--Best Practice Guidelines for Therapeutic Drug Monitoring: A Position Paper by the Subcommission on Therapeutic Drug Monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 2008, 49, 1239–1276. [Google Scholar] [CrossRef]
- Cotter, L.M.; Eadie, M.J.; Hooper, W.D.; Lander, C.M.; Smith, G.A.; Tyrer, J.H. The Pharmacokinetics of Carbamazepine. Eur. J. Clin. Pharmacol. 1977, 12, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Gérardin, A.P.; Abadie, F.V.; Campestrini, J.A.; Theobald, W. Pharmacokinetics of Carbamazepine in Normal Humans after Single and Repeated Oral Doses. J. Pharmacokinet. Biopharm. 1976, 4, 521–535. [Google Scholar] [CrossRef]
- FDA Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: http://Www.Fda.Gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/Ucm093664.Htm#4 (accessed on 23 May 2024).
- Greenberg, R.G.; Melloni, C.; Wu, H.; Gonzalez, D.; Ku, L.; Hill, K.D.; Hornik, C.P.; Cohen-Wolkowiez, M.; Guptill, J.T. Therapeutic Index Estimation of Antiepileptic Drugs: A Systematic Literature Review Approach. Clin. Neuropharmacol. 2016, 39, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Hayes, G.; Kootsikas, M.E. Reassessing the Lower End of the Phenytoin Therapeutic Range: A Review of the Literature. Ann. Pharmacother. 1993, 27, 1389–1392. [Google Scholar] [CrossRef]
- Cicali, B.; Lingineni, K.; Cristofoletti, R.; Wendl, T.; Hoechel, J.; Wiesinger, H.; Chaturvedula, A.; Vozmediano, V.; Schmidt, S. Quantitative Assessment of Levonorgestrel Binding Partner Interplay and Drug-Drug Interactions Using Physiologically Based Pharmacokinetic Modeling. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 48–58. [Google Scholar] [CrossRef]
- Wienkers, L.C.; Heath, T.G. Predicting in Vivo Drug Interactions from in Vitro Drug Discovery Data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Bolleddula, J.; Gopalakrishnan, S.; Hu, P.; Dong, J.; Venkatakrishnan, K. Alternatives to Rifampicin: A Review and Perspectives on the Choice of Strong CYP3A Inducers for Clinical Drug–Drug Interaction Studies. Clin. Transl. Sci. 2022, 15, 2075–2095. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration Physiologically Based Pharmacokinetic Analyses—Format and Content. Available online: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (accessed on 23 May 2024).
- EMA. Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation; European Medicines Agency: London, UK, 2018; Volume EMA/CHMP/4, ISBN EMA/CHMP/458101/2016. [Google Scholar]
- Food and Drug Administration, U.S. Guidance for Industry: Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. Available online: https://www.fda.gov/media/134582/download (accessed on 23 May 2024).
- Lukacova, V.; Parrott, N.J.; Lave, T.; Fraczkiewicz, G.; Bolger, M.B.; Woltosz, W.S. General Approach to Calculation of Tissue:Plasma Partition Coefficients for Physiologically Based Pharmacokinetic (PBPK) Modeling. In Proceedings of the AAPS Annual Meeting, Atlanta, GA, USA, 16–20 November 2008. [Google Scholar]
- Tomson, T.; Tybring, G.; Bertilsson, L. Single-Dose Kinetics and Metabolism of Carbamazepine-10,11-Epoxide. Clin. Pharmacol. Ther. 1983, 33, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Sumi, M.; Watari, N.; Umezawa, O.; Kaneniwa, N. Pharmacokinetic Study of Carbamazepine and Its Epoxide Metabolite in Humans. J. Pharmacobiodyn 1987, 10, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Spina, E.; Tomson, T.; Svensson, J.O.; Faigle, J.W.; Bertilsson, L. Single-Dose Kinetics of an Enteric-Coated Formulation of Carbamazepine-10,11-Epoxide, an Active Metabolite of Carbamazepine. Ther. Drug Monit. 1988, 10, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.F.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. The Human Intestinal Cytochrome P450 “Pie”. Drug Metab. Dispos. 2006, 34, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Kasteel, E.E.J.; Darney, K.; Kramer, N.I.; Dorne, J.L.C.M.; Lautz, L.S. Human Variability in Isoform-Specific UDP-Glucuronosyltransferases: Markers of Acute and Chronic Exposure, Polymorphisms and Uncertainty Factors. Arch. Toxicol. 2020, 94, 2637–2661. [Google Scholar] [CrossRef] [PubMed]
- Gérardin, A.; Dubois, J.P.; Moppert, J.; Geller, L. Absolute Bioavailability of Carbamazepine after Oral Administration of a 2% Syrup. Epilepsia 1990, 31, 334–338. [Google Scholar] [CrossRef]
- Meyer, M.C.; Straughn, A.B.; Jarvi, E.J.; Wood, G.C.; Pelsor, F.R.; Shah, V.P. The Bioinequivalence of Carbamazepine Tablets with a History of Clinical Failures. Pharm. Res. 1992, 9, 1612–1616. [Google Scholar] [CrossRef]
- Zhang, X.; Lionberger, R.A.; Davit, B.M.; Yu, L.X. Utility of Physiologically Based Absorption Modeling in Implementing Quality by Design in Drug Development. AAPS J. 2011, 13, 59–71. [Google Scholar] [CrossRef]
- Dam, M.; Christiansen, J.; Kristensen, C.B.; Helles, A.; Jaegerskou, A.; Schmiegelow, M. Carbamazepine: A Clinical Biopharmaceutical Study. Eur. J. Clin. Pharmacol. 1981, 20, 59–64. [Google Scholar] [CrossRef] [PubMed]
- El-Massik, M.A.; Abdallah, O.Y.; Galal, S.; Daabis, N.A. Towards a Universal Dissolution Medium for Carbamazepine. Drug Dev. Ind. Pharm. 2006, 32, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, P.; Lacasse, F.-X.; Preda, M.; Thibert, R.; Clas, S.-D.; McMullen, J.N. Physicochemical Evaluation of Carbamazepine Microparticles Produced by the Rapid Expansion of Supercritical Solutions and by Spray-Drying. Pharm. Dev. Technol. 2003, 8, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Damle, B.; Xie, J.; Unger, S.E.; Grasela, D.M.; Kaul, S. Pharmacokinetic Interaction between Efavirenz and Carbamazepine after Multiple-Dose Administration in Healthy Subjects. J. Clin. Pharmacol. 2008, 48, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.G.; Patel, R.; Clark, R.J.; Ho, T.; Trisdale, S.K.; Fang, Y.; Stresser, D.M. Effect of Fifteen CYP3A4 in Vitro Inducers on the Induction of Hepatocytes: A Trend Analysis. In Proceedings of the 20th North American ISSX Meeting, Orlando FL, USA, 18–22 October 2015; p. 2015. [Google Scholar]
- Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug–Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics 2021, 13, 270. [Google Scholar] [CrossRef] [PubMed]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK Models for CYP3A4 and P-Gp DDI Prediction: A Modeling Network of Rifampicin, Itraconazole, Clarithromycin, Midazolam, Alfentanil, and Digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef]
- Rodriguez-Vera, L.; Yin, X.; Almoslem, M.; Romahn, K.; Cicali, B.; Lukacova, V.; Cristofoletti, R.; Schmidt, S. Comprehensive Physiologically Based Pharmacokinetic Model to Assess Drug-Drug Interactions of Phenytoin. Pharmaceutics 2023, 15, 2486. [Google Scholar] [CrossRef] [PubMed]
- Guest, E.J.; Aarons, L.; Houston, J.B.; Rostami-Hodjegan, A.; Galetin, A. Critique of the Two-Fold Measure of Prediction Success for Ratios: Application for the Assessment of Drug-Drug Interactions. Drug Metab. Dispos. 2011, 39, 170–173. [Google Scholar] [CrossRef]
- Scheytt, T.; Mersmann, P.; Lindstädt, R.; Heberer, T. 1-Octanol/Water Partition Coefficients of 5 Pharmaceuticals from Human Medical Care: Carbamazepine, Clofibric Acid, Diclofenac, Ibuprofen, and Propyphenazone. Water Air Soil. Pollut. 2005, 165, 3–11. [Google Scholar] [CrossRef]
- Zhu, C.; Jiang, L.; Chen, T.-M.; Hwang, K.-K. A Comparative Study of Artificial Membrane Permeability Assay for High Throughput Profiling of Drug Absorption Potential. Eur. J. Med. Chem. 2002, 37, 399–407. [Google Scholar] [CrossRef]
- Lombardo, F.; Shalaeva, M.Y.; Tupper, K.A.; Gao, F.; Abraham, M.H. ElogPoct: A Tool for Lipophilicity Determination in Drug Discovery. J. Med. Chem. 2000, 43, 2922–2928. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Ahman, M.; Holmén, A.G. Relationship between Brain Tissue Partitioning and Microemulsion Retention Factors of CNS Drugs. J. Med. Chem. 2009, 52, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular Properties of WHO Essential Drugs and Provisional Biopharmaceutical Classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.; Williams, K. Metabolism and Pharmacokinetics of Ibuprofen. In Aspirin and Related Drugs; CRC Press: Boca Raton, FL, USA, 2004; Volume 4, pp. 157–180. ISBN 9780203646960. [Google Scholar]
- Bonneton, J.; Genton, P.; Mesdjian, E. Distribution of Carbamazepine and Its Epoxide in Blood Compartments in Adolescent and Adult Epileptic Patients. Biopharm. Drug Dispos. 1992, 13, 411–416. [Google Scholar] [CrossRef] [PubMed]
- de Groot, G.; van Heijst, A.N.P.; Maes, R.A.A. Charcoal Hemoperfusion in the Treatment of Two Cases of Acute Carbamazepine Poisoning. J. Toxicol. Clin. Toxicol. 1984, 22, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Kovaĉević, I.; Parojĉić, J.; Homŝek, I.; Tubić-Grozdanis, M.; Langguth, P. Justification of Biowaiver for Carbamazepine, a Low Soluble High Permeable Compound, in Solid Dosage Forms Based on IVIVC and Gastrointestinal Simulation. Mol. Pharm. 2009, 6, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Cazali, N.; Tran, A.; Treluyer, J.M.; Rey, E.; D’Athis, P.; Vincent, J.; Pons, G. Inhibitory Effect of Stiripentol on Carbamazepine and Saquinavir Metabolism in Human. Br. J. Clin. Pharmacol. 2003, 56, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Henshall, J.; Galetin, A.; Harrison, A.; Houston, J.B. Comparative Analysis of CYP3A Heteroactivation by Steroid Hormones and Flavonoids in Different in Vitro Systems and Potential in Vivo Implications. Drug Metab. Dispos. 2008, 36, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Lennernäs, H. Intestinal Permeability and Its Relevance for Absorption and Elimination. Xenobiotica 2007, 37, 1015–1051. [Google Scholar] [CrossRef]
- Winiwarter, S.; Bonham, N.M.; Ax, F.; Hallberg, A.; Lennernäs, H.; Karlén, A. Correlation of Human Jejunal Permeability (in Vivo) of Drugs with Experimentally and Theoretically Derived Parameters. A Multivariate Data Analysis Approach. J. Med. Chem. 1998, 41, 4939–4949. [Google Scholar] [CrossRef]
- Söderlind, E.; Karlsson, E.; Carlsson, A.; Kong, R.; Lenz, A.; Lindborg, S.; Sheng, J.J. Simulating Fasted Human Intestinal Fluids: Understanding the Roles of Lecithin and Bile Acids. Mol. Pharm. 2010, 7, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, P.K.; Suresh, B.; Hussaini, S.S.Q.; Rao, Y.M.; Apte, S. Comparative in Vitro Study of Six Carbamazepine Products. AAPS Pharm. Sci. Tech. 2008, 9, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Kohlmann, P.; Stillhart, C.; Kuentz, M.; Parrott, N. Investigating Oral Absorption of Carbamazepine in Pediatric Populations. AAPS J. 2017, 19, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Clarysse, S.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Intestinal Drug Solubility Estimation Based on Simulated Intestinal Fluids: Comparison with Solubility in Human Intestinal Fluids. Eur. J. Pharm. Sci. 2011, 43, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.; Knox, C.; Guo, A.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. Drugbank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668–D672. Available online: https://go.drugbank.com/metabolites/DBMET00291 (accessed on 23 May 2024). [CrossRef] [PubMed]
- Shahzadi, A.; Javed, I.; Aslam, B.; Muhammad, F.; Asi, M.R.; Ashraf, M.Y. Zia-ur-Rahman Therapeutic Effects of Ciprofloxacin on the Pharmacokinetics of Carbamazepine in Healthy Adult Male Volunteers. Pak. J. Pharm. Sci. 2011, 24, 63–68. [Google Scholar] [PubMed]
- Shou, M.; Hayashi, M.; Pan, Y.; Xu, Y.; Morrissey, K.; Xu, L.; Skiles, G.L. Modeling, Prediction, and in Vitro in Vivo Correlation of CYP3A4 Induction. Drug Metab. Dispos. 2008, 36, 2355–2370. [Google Scholar] [CrossRef] [PubMed]
- Almond, L.M.; Mukadam, S.; Gardner, I.; Okialda, K.; Wong, S.; Hatley, O.; Tay, S.; Rowland-Yeo, K.; Jamei, M.; Rostami-Hodjegan, A.; et al. Prediction of Drug-Drug Interactions Arising from CYP3A Induction Using a Physiologically Based Dynamic Models. Drug Metab. Dispos. 2016, 44, 821–832. [Google Scholar] [CrossRef]
- Zhang, J.G.; Ho, T.; Callendrello, A.L.; Clark, R.J.; Santone, E.A.; Kinsman, S.; Xiao, D.; Fox, L.G.; Einolf, H.J.; Stresser, D.M. Evaluation of Calibration Curve-Based Approaches to Predict Clinical Inducers and Noninducers of CYP3A4 with Plated Human Hepatocytes. Drug Metab. Dispos. 2014, 42, 1379–1391. [Google Scholar] [CrossRef]
- McGinnity, D.F.; Zhang, G.; Kenny, J.R.; Hamilton, G.A.; Otmani, S.; Stams, K.R.; Haney, S.; Brassil, P.; Stresser, D.M.; Riley, R.J. Evaluation of Multiple in Vitro Systems for Assessment of CYP3A4 Induction in Drug Discovery: Human Hepatocytes, Pregnane X Receptor Reporter Gene, and Fa2N-4 and HepaRG Cells. Drug Metab. Dispos. 2009, 37, 1259–1268. [Google Scholar] [CrossRef]
- Moore, A.; Chothe, P.P.; Tsao, H.; Hariparsad, N. Evaluation of the Interplay between Uptake Transport and CYP3A4 Induction in Micropatterned Cocultured Hepatocytes. Drug Metab. Dispos. 2016, 44, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Savaryn, J.P.; Sun, J.; Ma, J.; Jenkins, G.J.; Stresser, D.M. Broad Application of CYP3A4 Liquid Chromatography-Mass Spectrometry Protein Quantification in Hepatocyte Cytochrome P450 Induction Assays Identifies Nonuniformity in MRNA and Protein Induction Responses. Drug Metab. Dispos. 2022, 50, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, O.A.; Kish, M.; Boldt, S.; Obach, R.S. Cytochrome P450 3A4 MRNA Is a More Reliable Marker than CYP3A4 Activity for Detecting Pregnane X Receptor-Activated Induction of Drug-Metabolizing Enzymes. Drug Metab. Dispos. 2010, 38, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Vermet, H.; Raoust, N.; Ngo, R.; Esserméant, L.; Klieber, S.; Fabre, G.; Boulenc, X. Evaluation of Normalization Methods To Predict CYP3A4 Induction in Six Fully Characterized Cryopreserved Human Hepatocyte Preparations and HepaRG Cells. Drug Metab. Dispos. 2016, 44, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chothe, P.P.; Sager, J.E.; Tsao, H.; Moore, A.; Laitinen, L.; Hariparsad, N. Quantitative Prediction of CYP3A4 Induction: Impact of Measured, Free, and Intracellular Perpetrator Concentrations from Human Hepatocyte Induction Studies on Drug-Drug Interaction Predictions. Drug Metab. Dispos. 2017, 45, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, I.; Murayama, N.; Kuroki, A.; Kota, J.; Iwano, S.; Yamazaki, H.; Hirota, T. Evaluation of Cytochrome P450 Inductions by Anti-Epileptic Drug Oxcarbazepine, 10-Hydroxyoxcarbazepine, and Carbamazepine Using Human Hepatocytes and HepaRG Cells. Xenobiotica 2016, 46, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, O.A.; Boldt, S.; Kish, M.; Obach, R.S.; Tremaine, L.M. Prediction of Drug-Drug Interactions from in Vitro Induction Data: Application of the Relative Induction Score Approach Using Cryopreserved Human Hepatocytes. Drug Metab. Dispos. 2008, 36, 1971–1974. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, S.; Kato, M.; Shindoh, H.; Kaneko, A.; Ishigai, M.; Miyauchi, S. Simple Evaluation Method for CYP3A4 Induction from Human Hepatocytes: The Relative Factor Approach with an Induction Detection Limit Concentration Based on the Emax Model. Drug Metab. Dispos. 2017, 45, 1139–1145. [Google Scholar] [CrossRef]
- Usui, T.; Saitoh, Y.; Komada, F. Induction of CYP3As in HepG2 Cells by Several Drugs. Association between Induction of CYP3A4 and Expression of Glucocorticoid Receptor. Biol. Pharm. Bull. 2003, 26, 510–517. [Google Scholar] [CrossRef]
- Nagai, M.; Hosaka, T.; Satsukawa, M.; Yoshinari, K. Characterization of CYP2C Induction in Cryopreserved Human Hepatocytes and Its Application in the Prediction of the Clinical Consequences of the Induction. J. Pharm. Sci. 2018, 107, 2479–2488. [Google Scholar] [CrossRef]
- Dickmann, L.J.; Isoherranen, N. Quantitative Prediction of CYP2B6 Induction by Estradiol during Pregnancy: Potential Explanation for Increased Methadone Clearance during Pregnancy. Drug Metab. Dispos. 2013, 41, 270–274. [Google Scholar] [CrossRef]
- Faucette, S.R.; Wang, H.; Hamilton, G.A.; Jolley, S.L.; Gilbert, D.; Lindley, C.; Yan, B.; Negishi, M.; LeCluyse, E.L. Regulation of CYP2B6 in Primary Human Hepatocytes by Prototypical Inducers. Drug Metab. Dispos. 2004, 32, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Sahi, J.; Shord, S.S.; Lindley, C.; Ferguson, S.; LeCluyse, E.L. Regulation of Cytochrome P450 2C9 Expression in Primary Cultures of Human Hepatocytes. J. Biochem. Mol. Toxicol. 2009, 23, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, A.-H.; Brøsen, K.; Damkier, P. A Comparative Pharmacokinetic Study in Healthy Volunteers of the Effect of Carbamazepine and Oxcarbazepine on Cyp3a4. Epilepsia 2007, 48, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Song, I.; Weller, S.; Patel, J.; Borland, J.; Wynne, B.; Choukour, M.; Jerva, F.; Piscitelli, S. Effect of Carbamazepine on Dolutegravir Pharmacokinetics and Dosing Recommendation. Eur. J. Clin. Pharmacol. 2016, 72, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.L.; Lin, T.S.; Huang, J.D. Effect of Single- and Multiple-Dose Carbamazepine on the Pharmacokinetics of Diphenylhydantoin. Eur. J. Clin. Pharmacol. 1992, 43, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Tomson, T.; Tybring, G.; Bertilsson, L. Carbamazepine Metabolism in Man. Induction and Pharmacogenetic Aspects. Clin. Pharmacokinet. 1985, 10, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yun, W.; Sun, X.; Qiu, F.; Zhao, L.; Guo, Y. Effects of Major Transporter and Metabolizing Enzyme Gene Polymorphisms on Carbamazepine Metabolism in Chinese Patients with Epilepsy. Pharmacogenomics 2014, 15, 1867–1879. [Google Scholar] [CrossRef]
- Zhao, G.-X.; Zhang, Z.; Cai, W.-K.; Shen, M.-L.; Wang, P.; He, G.-H. Associations between CYP3A4, CYP3A5 and SCN1A Polymorphisms and Carbamazepine Metabolism in Epilepsy: A Meta-Analysis. Epilepsy Res. 2021, 173, 106615. [Google Scholar] [CrossRef]
- Ma, C.-L.; Jiao, Z.; Wu, X.-Y.; Hong, Z.; Wu, Z.-Y.; Zhong, M.-K. Association between PK/PD-Involved Gene Polymorphisms and Carbamazepine-Individualized Therapy. Pharmacogenomics 2015, 16, 1499–1512. [Google Scholar] [CrossRef]
- Miyata-Nozaka, Y.; Zain, S.M.; Taguchi, M.; Shigeyama, M.; Isobe, T.; Hanioka, N. Carbamazepine 10,11-Epoxidation in Human Liver Microsomes: Influence of the CYP3A5*3 Polymorphism. Pharmazie 2017, 72, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Seo, T.; Nakada, N.; Ueda, N.; Hagiwara, T.; Hashimoto, N.; Nakagawa, K.; Ishitsu, T. Effect of CYP3A5*3 on Carbamazepine Pharmacokinetics in Japanese Patients with Epilepsy. Clin. Pharmacol. Ther. 2006, 79, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Park, P.W.; Seo, Y.H.; Ahn, J.Y.; Kim, K.A.; Park, J.Y. Effect of CYP3A5*3 Genotype on Serum Carbamazepine Concentrations at Steady-State in Korean Epileptic Patients. J. Clin. Pharm. Ther. 2009, 34, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Saito, Y.; Shiseki, K.; Fukushima-Uesaka, H.; Hasegawa, R.; Ozawa, S.; Sugai, K.; Katoh, M.; Saitoh, O.; Ohnuma, T.; et al. Haplotype Structures of EPHX1 and Their Effects on the Metabolism of Carbamazepine-10,11-Epoxide in Japanese Epileptic Patients. Eur. J. Clin. Pharmacol. 2005, 61, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, K.; Itoda, M.; Hanioka, N.; Saito, Y.; Murayama, N.; Nakajima, O.; Soyama, A.; Ishida, S.; Ozawa, S.; Ando, M.; et al. Non-Synonymous Single Nucleotide Alterations in the Microsomal Epoxide Hydrolase Gene and Their Functional Effects. Xenobiotica 2003, 33, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-L.; Chen, X.-L.; Bai, Z.-F.; Zhao, X.; Li, W.-X.; Wang, X.-Y.; Zhang, H.; Chen, X.-F.; Zhang, S.-Q.; Tang, J.-F.; et al. ABCB1 c.3435C > T and EPHX1 c.416A > G Polymorphisms Influence Plasma Carbamazepine Concentration, Metabolism, and Pharmacoresistance in Epileptic Patients. Gene 2021, 805, 145907. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-C.; Chang, W.-L.; Ho, J.-L.; Tai, J.J.; Hsieh, T.-J.; Huang, H.-C.; Hsieh, Y.-W.; Liou, H.-H. Association of Polymorphisms in EPHX1, UGT2B7, ABCB1, ABCC2, SCN1A and SCN2A Genes with Carbamazepine Therapy Optimization. Pharmacogenomics 2012, 13, 159–169. [Google Scholar] [CrossRef]
- Zhang, C.; Zuo, Z.; Kwan, P.; Baum, L. In Vitro Transport Profile of Carbamazepine, Oxcarbazepine, Eslicarbazepine Acetate, and Their Active Metabolites by Human P-Glycoprotein. Epilepsia 2011, 52, 1894–1904. [Google Scholar] [CrossRef]
- Mohutsky, M.A.; Romeike, A.; Meador, V.; Lee, W.M.; Fowler, J.; Francke-Carroll, S. Hepatic Drug-Metabolizing Enzyme Induction and Implications for Preclinical and Clinical Risk Assessment. Toxicol. Pathol. 2010, 38, 799–809. [Google Scholar] [CrossRef]
- Ekiciler, A.; Chen, W.L.K.; Bo, Y.; Pugliano, A.; Donzelli, M.; Parrott, N.; Umehara, K. Quantitative Cytochrome P450 3A4 Induction Risk Assessment Using Human Hepatocytes Complemented with Pregnane X Receptor-Activating Profiles. Drug Metab. Dispos. 2023, 51, 276–284. [Google Scholar] [CrossRef]
- Hariparsad, N.; Ramsden, D.; Palamanda, J.; Dekeyser, J.G.; Fahmi, O.A.; Kenny, J.R.; Einolf, H.; Mohutsky, M.; Pardon, M.; Siu, Y.A.; et al. Considerations from the IQ Induction Working Group in Response to Drug-Drug Interaction Guidance from Regulatory Agencies: Focus on Downregulation, CYP2C Induction, and CYP2B6 Positive Control. Drug Metab. Dispos. 2017, 45, 1049–1059. [Google Scholar] [CrossRef]
- Lin, Y.S.; Dowling, A.L.S.; Quigley, S.D.; Farin, F.M.; Zhang, J.; Lamba, J.; Schuetz, E.G.; Thummel, K.E. Co-Regulation of CYP3A4 and CYP3A5 and Contribution to Hepatic and Intestinal Midazolam Metabolism. Mol. Pharmacol. 2002, 62, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; et al. Sequence Diversity in CYP3A Promoters and Characterization of the Genetic Basis of Polymorphic CYP3A5 Expression. Nat. Genet. 2001, 27, 383–391. [Google Scholar] [CrossRef]
- Gorski, J.C.; Hall, S.D.; Jones, D.R.; VandenBranden, M.; Wrighton, S.A. Regioselective Biotransformation of Midazolam by Members of the Human Cytochrome P450 3A (CYP3A) Subfamily. Biochem. Pharmacol. 1994, 47, 1643–1653. [Google Scholar] [CrossRef]
- Oscarson, M.; Zanger, U.M.; Rifki, O.F.; Klein, K.; Eichelbaum, M.; Meyer, U.A. Transcriptional Profiling of Genes Induced in the Livers of Patients Treated with Carbamazepine. Clin. Pharmacol. Ther. 2006, 80, 440–456. [Google Scholar] [CrossRef]
- Järvinen, E.; Hammer, H.S.; Pötz, O.; Ingelman-Sundberg, M.; Stage, T.B. 3D Spheroid Primary Human Hepatocytes for Prediction of Cytochrome P450 and Drug Transporter Induction. Clin. Pharmacol. Ther. 2023, 113, 1284–1294. [Google Scholar] [CrossRef]
- Owen, A.; Pirmohamed, M.; Tettey, J.N.; Morgan, P.; Chadwick, D.; Park, B.K. Carbamazepine Is Not a Substrate for P-Glycoprotein. Br. J. Clin. Pharmacol. 2001, 51, 345–349. [Google Scholar] [CrossRef]
- Potschka, H.; Fedrowitz, M.; Löscher, W. P-Glycoprotein and Multidrug Resistance-Associated Protein Are Involved in the Regulation of Extracellular Levels of the Major Antiepileptic Drug Carbamazepine in the Brain. Neuroreport 2001, 12, 3557–3560. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Rodrigues, M.; Meirinho, S.; Fortuna, A.; Falcão, A.; Alves, G. Silymarin as a Flavonoid-Type P-Glycoprotein Inhibitor with Impact on the Pharmacokinetics of Carbamazepine, Oxcarbazepine and Phenytoin in Rats. Drug Chem. Toxicol. 2021, 44, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Rädisch, S.; Dickens, D.; Lang, T.; Bonnett, L.; Arlanov, R.; Johnson, M.R.; Schwab, M.; Marson, A.G.; Pirmohamed, M. A Comprehensive Functional and Clinical Analysis of ABCC2 and Its Impact on Treatment Response to Carbamazepine. Pharmacogenomics J. 2014, 14, 481–487. [Google Scholar] [CrossRef]
- Awasthi, S.; Hallene, K.L.; Fazio, V.; Singhal, S.S.; Cucullo, L.; Awasthi, Y.C.; Dini, G.; Janigro, D. RLIP76, a Non-ABC Transporter, and Drug Resistance in Epilepsy. BMC Neurosci. 2005, 6, 61. [Google Scholar] [CrossRef]
- Giessmann, T.; May, K.; Modess, C.; Wegner, D.; Hecker, U.; Zschiesche, M.; Dazert, P.; Grube, M.; Schroeder, E.; Warzok, R.; et al. Carbamazepine Regulates Intestinal P-Glycoprotein and Multidrug Resistance Protein MRP2 and Influences Disposition of Talinolol in Humans. Clin. Pharmacol. Ther. 2004, 76, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Brueck, S.; Bruckmueller, H.; Wegner, D.; Busch, D.; Martin, P.; Oswald, S.; Cascorbi, I.; Siegmund, W. Transcriptional and Post-Transcriptional Regulation of Duodenal P-Glycoprotein and MRP2 in Healthy Human Subjects after Chronic Treatment with Rifampin and Carbamazepine. Mol. Pharm. 2019, 16, 3823–3830. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ke, M.; Fang, W.; Jiang, Y.; Lin, R.; Wu, W.; Huang, P.; Lin, C. Physiologically Based Pharmacokinetic Modeling to Predict Maternal Pharmacokinetics and Fetal Carbamazepine Exposure during Pregnancy. Eur. J. Pharm. Sci. 2024, 194, 106707. [Google Scholar] [CrossRef]
- Conner, T.M.; Nikolian, V.C.; Georgoff, P.E.; Pai, M.P.; Alam, H.B.; Sun, D.; Reed, R.C.; Zhang, T. Physiologically Based Pharmacokinetic Modeling of Disposition and Drug-Drug Interactions for Valproic Acid and Divalproex. Eur. J. Pharm. Sci. 2018, 111, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Schuck, E.; Ferry, J.; Gidal, B.; Hussein, Z. Changes in Perampanel Levels during De-Induction: Simulations Following Carbamazepine Discontinuation. Acta Neurol. Scand. 2020, 142, 131–138. [Google Scholar] [CrossRef]
- Ngo, L.T.; Yang, S.-Y.; Shin, S.; Cao, D.T.; Van Nguyen, H.; Jung, S.; Lee, J.-Y.; Lee, J.-H.; Yun, H.-Y.; Chae, J.-W. Application of Physiologically-Based Pharmacokinetic Model Approach to Predict Pharmacokinetics and Drug-Drug Interaction of Rivaroxaban: A Case Study of Rivaroxaban and Carbamazepine. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Marok, F.Z.; Fuhr, L.M.; Hanke, N.; Selzer, D.; Lehr, T. Physiologically Based Pharmacokinetic Modeling of Bupropion and Its Metabolites in a CYP2B6 Drug-Drug-Gene Interaction Network. Pharmaceutics 2021, 13, 331. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, X.; Cicali, B.; Rodriguez-Vera, L.; Lukacova, V.; Cristofoletti, R.; Schmidt, S. Applying Physiologically Based Pharmacokinetic Modeling to Interpret Carbamazepine’s Nonlinear Pharmacokinetics and Its Induction Potential on Cytochrome P450 3A4 and Cytochrome P450 2C9 Enzymes. Pharmaceutics 2024, 16, 737. https://doi.org/10.3390/pharmaceutics16060737
Yin X, Cicali B, Rodriguez-Vera L, Lukacova V, Cristofoletti R, Schmidt S. Applying Physiologically Based Pharmacokinetic Modeling to Interpret Carbamazepine’s Nonlinear Pharmacokinetics and Its Induction Potential on Cytochrome P450 3A4 and Cytochrome P450 2C9 Enzymes. Pharmaceutics. 2024; 16(6):737. https://doi.org/10.3390/pharmaceutics16060737
Chicago/Turabian StyleYin, Xuefen, Brian Cicali, Leyanis Rodriguez-Vera, Viera Lukacova, Rodrigo Cristofoletti, and Stephan Schmidt. 2024. "Applying Physiologically Based Pharmacokinetic Modeling to Interpret Carbamazepine’s Nonlinear Pharmacokinetics and Its Induction Potential on Cytochrome P450 3A4 and Cytochrome P450 2C9 Enzymes" Pharmaceutics 16, no. 6: 737. https://doi.org/10.3390/pharmaceutics16060737
APA StyleYin, X., Cicali, B., Rodriguez-Vera, L., Lukacova, V., Cristofoletti, R., & Schmidt, S. (2024). Applying Physiologically Based Pharmacokinetic Modeling to Interpret Carbamazepine’s Nonlinear Pharmacokinetics and Its Induction Potential on Cytochrome P450 3A4 and Cytochrome P450 2C9 Enzymes. Pharmaceutics, 16(6), 737. https://doi.org/10.3390/pharmaceutics16060737