Physiologically Based Pharmacokinetic Modeling in Pregnancy, during Lactation and in Neonates: Achievements, Challenges and Future Directions



{kind=link}
1. Introduction
2. An Overview of Published Articles
2.1. Pregnancy-Related Physiologically Based Pharmacokinetics Papers
2.2. Neonatal Physiologically Based Pharmacokinetics Papers
2.3. Lactation-Related Physiologically Based Pharmacokinetic Papers
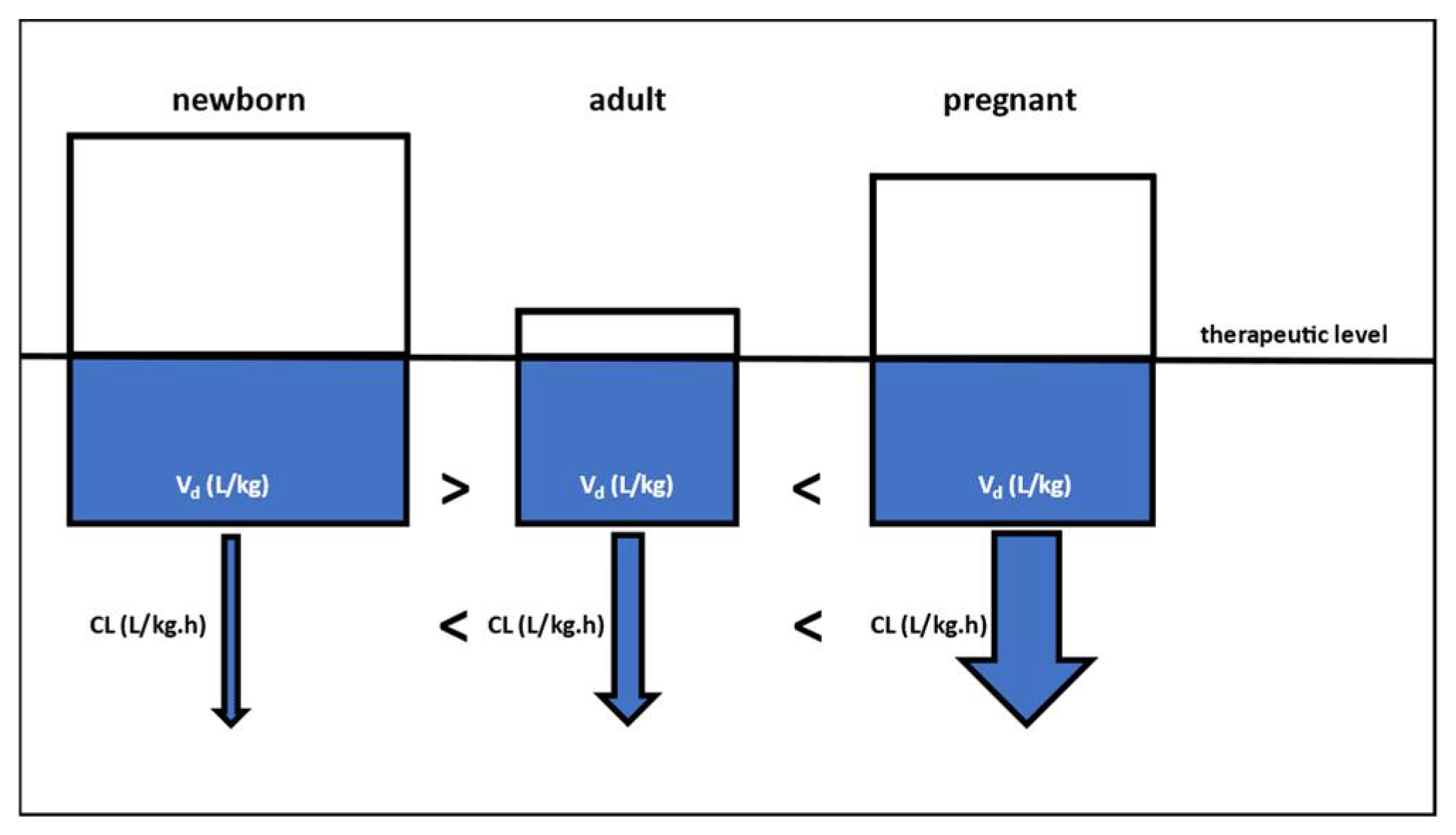
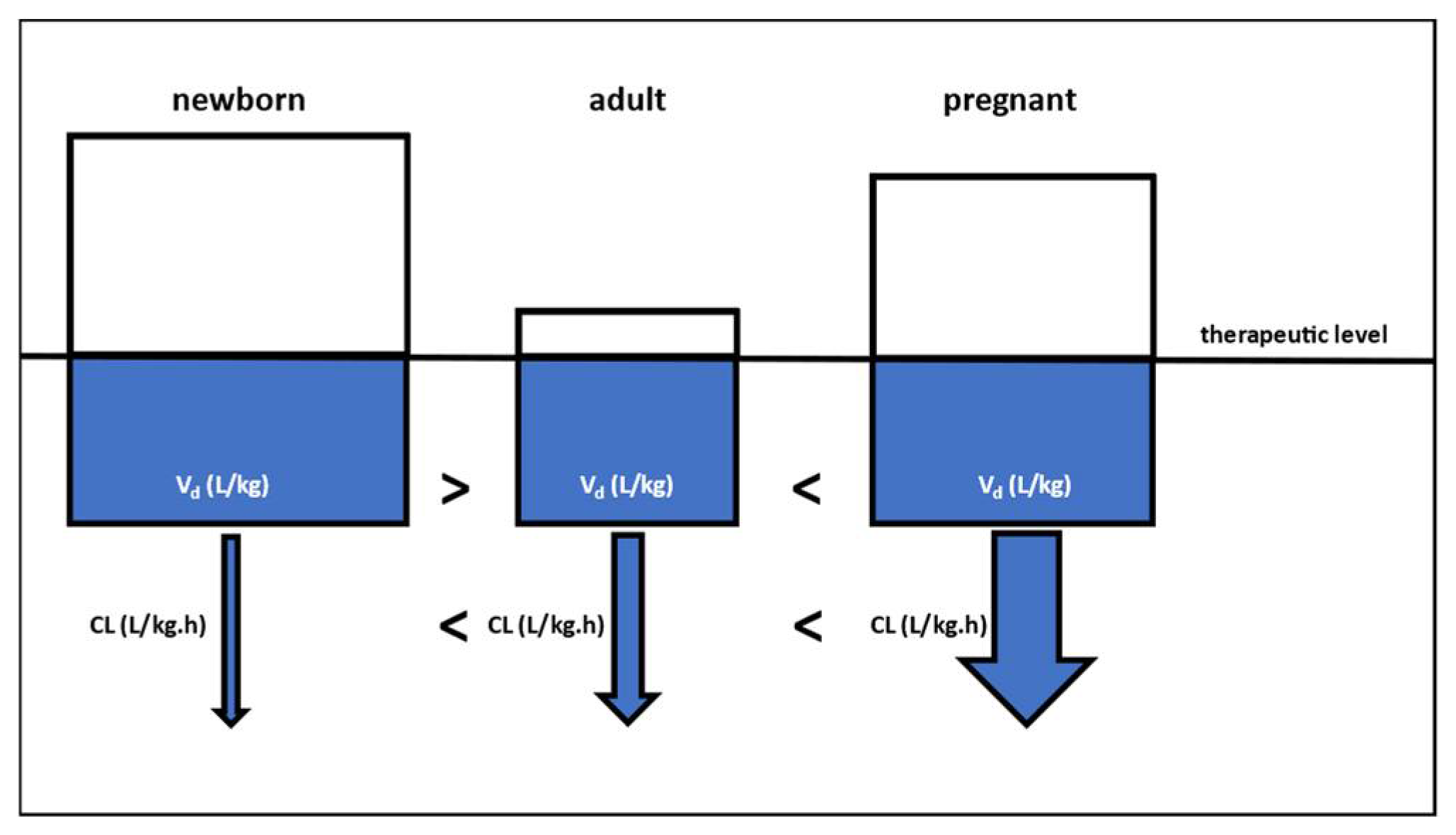
3. Lessons Learned and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
List of Contributions
- Le Merdy, M.; Szeto, K.X.; Perrier, J.; Bolger, M.B.; Lukacova, V. PBPK Modeling Approach to Predict the Behavior of Drugs Cleared by Metabolism in Pregnant Subjects and Fetuses. Pharmaceutics 2024, 16, 96. https://doi.org/10.3390/pharmaceutics16010096.
- Gong, C.; Bertagnolli, L.N.; Boulton, D.W.; Coppola, P. A Literature Review of Changes in Phase II Drug-Metabolizing Enzyme and Drug Transporter Expression during Pregnancy. Pharmaceutics 2023, 15, 2624. https://doi.org/10.3390/pharmaceutics15112624.
- Coppola, P.; Butler, A.; Cole, S.; Kerwash, E. Total and Free Blood and Plasma Concentration Changes in Pregnancy for Medicines Highly Bound to Plasma Proteins: Application of Physiologically Based Pharmacokinetic Modelling to Understand the Impact on Efficacy. Pharmaceutics 2023, 15, 2455. https://doi.org/10.3390/pharmaceutics15102455.
- Abduljalil, K.; Gardner, I.; Jamei, M. An Application of a Physiologically Based Pharmacokinetic Approach to Predict Ceftazidime Pharmacokinetics in a Pregnant Population. Pharmaceutics 2024, 16, 474.
- Van Hoogdalem, M.W.; Tanaka, R.; Abduljalil, K.; Jonson, T.N.; Wexelblatt, S.L.; Akinbi, H.T.; Vinks, A.A.; Mizuno, T. Forecasting Fetal Buprenorphine Exposure through Maternal-Fetal Physiologically Based Pharmacokinetic Modeling. Pharmaceutics 2024, 16, 375. https://doi.org/10.3390/pharmaceutics16030375.
- Yang, X.; Grimstein, M.; Pressly, M.; Fletcher, E.P.; Shord, S.; Leong, R. Utility of Physiologically Based Pharmacokinetic Modeling to Investigate the Impact of Physiological Changes of Pregnancy and Cancer on Oncology Drug Pharmacokinetics. Pharmaceutics 2023, 15, 2727. https://doi.org/10.3390/pharmaceutics15122727.
- Dinh, J.; Johnson, T.N.; Grimstein, M.; Lewis, T. Physiologically Based Pharmacokinetics Modeling in the Neonatal Population—Current Advances, Challenges, and Opportunities. Pharmaceutics 2023, 15, 2579. https://doi.org/10.3390/pharmaceutics15112579.
- Zhang, W.; Zhang, Q.; Cao, Z.; Zheng, L.; Hu, W. Physiologically Based Pharmacokinetic Modeling in Neonates: Current Status and Future Perspectives. Pharmaceutics 2023, 15, 2765. https://doi.org/10.3390/pharmaceutics15122765.
- De Sutter, P.-J.; Rossignol, P.; Breëns, L.; Gasthuys, E.; Vermeulen, A. Predicting Volume of Distribution in Neonates: Performance of Physiologically Based Pharmacokinetic Modelling. Pharmaceutics 2023, 15, 2348. https://doi.org/10.3390/pharmaceutics15092348.
- Nauwelaerts, N.; Macente, J.; Deferm, N.; Bonan, R.H.; Huang, M.-C.; Van Neste, M.; Bibi, D.; Badee, J.; Martins, F.S.; Smits, A.; et al. Generic Workflow to Predict Medicine Concentrations in Human Milk Using Physiologically-Based Pharmacokinetic (PBPK) Modelling—A Contribution from the ConcePTION Project. Pharmaceutics 2023, 15, 1469. https://doi.org/10.3390/pharmaceutics15051469.
- Shenkoya, B.; Yellepeddi, V.; Mark, K.; Gopalakrishnan, M. Predicting Maternal and Infant Tetrahydrocannabinol Exposure in Lactating Cannabis Users: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics 2023, 15, 2467. https://doi.org/10.3390/pharmaceutics15102467.
- Van Neste, M.; Bogaerts, A.; Nauwelaerts, N.; Macente, J.; Smits, A.; Annaert, P.; Allegaert, K. Challenges Related to Acquisition of Physiological Data for Physiologically Based Pharmacokinetic (PBPK) Models in Postpartum, Lactating Women and Breastfed Infants—A Contribution from the ConcePTION Project. Pharmaceutics 2023, 15, 2618. https://doi.org/10.3390/pharmaceutics15112618.
References
- Quinney, S.K.; Bies, R.R.; Grannis, S.J.; Bartlett, C.W.; Mendonca, E.; Rogerson, C.M.; Backes, C.H.; Shah, D.K.; Tillman, E.M.; Costantine, M.M.; et al. The MPRINT Hub Data, Model, Knowledge and Research Coordination Center: Bridging the gap in maternal–pediatric therapeutics research through data integration and pharmacometrics. Pharmacotherapy 2023, 43, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Kazma, J.M.; van den Anker, J.; Allegaert, K.; Dallmann, A.; Ahmadzia, H.K. Anatomical and physiological alterations of pregnancy. J. Pharmacokinet. Pharmacodyn. 2020, 47, 271–285. [Google Scholar] [CrossRef]
- Nooney, J.; Thor, S.; de Vries, C.; Clements, J.; Sahin, L.; Hua, W.; Everett, D.; Zaccaria, C.; Ball, R.; Saint-Raymond, A.; et al. Assuring access to safe medicines in pregnancy and breastfeeding. Clin. Pharmacol. Ther. 2021, 110, 941–945. [Google Scholar] [CrossRef]
- Eke, A.C.; Gebreyohannes, R.D.; Scantamburlo Fernandes, M.F. Physiologic changes during pregnancy and impact on small-molecule drugs, biological (monoclonal antibody) disposition, and response. J. Clin. Pharmacol. 2023, 63 (Suppl. S1), S34–S50. [Google Scholar] [CrossRef] [PubMed]
- Dallmann, A.; Mian, P.; van den Anker, J.; Allegaert, K. Clinical pharmacokinetic studies in pregnant women and the relevance of pharmacometrics tools. Curr. Pharm. Des. 2019, 25, 483–495. [Google Scholar] [CrossRef]
- Lin, W.; Chen, Y.; Unadkat, J.D.; Zhang, X.; Wu, D.; Heimbach, T. Applications, challenges, and outlook of PBPK modeling and simulation: A regulatory, industrial and academic perspective. Pharm. Res. 2022, 39, 1701–1731. [Google Scholar] [CrossRef] [PubMed]
- Szeto, K.X.; Le Merdy, M.; Dupont, B.; Bolger, M.B.; Lukacova, V. PBPK modeling approach to predict the behavior of drugs cleared by kidney in pregnant subjects and fetus. AAPS J. 2021, 23, 89. [Google Scholar] [CrossRef] [PubMed]
- Berezhkovskiy, L.M. Volume of Distribution at Steady State for a Linear Pharmacokinetic System with Peripheral Elimination. J. Pharm. Sci. 2004, 93, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Mechanistic Approaches to Volume of Distribution Predictions: Understanding the Processes. Pharm. Res. 2007, 24, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Allegaert, K.; Smits, A.; Annaert, P. Interdisciplinary Collaboration on Real World Data to Close the Knowledge Gap: A Reflection on “De Sutter et al. Predicting Volume of Distribution in Neonates: Performance of Physiologically Based Pharmacokinetic Modelling”. Pharmaceutics 2024, 16, 128. [Google Scholar] [CrossRef] [PubMed]
- Fashe, M.M.; Miner, T.A.; Fallon, J.K.; Schauer, A.P.; Sykes, C.; Smith, P.C.; Lee, C.R. Pregnancy related hormones increase CYP3A mediated buprenorphine metabolism in human hepatocytes: A comparison to CYP3A substrates nifedipine and midazolam. Front. Pharmacol. 2023, 14, 1218703. [Google Scholar] [CrossRef] [PubMed]
- Hebert, M.F.; Easterling, T.R.; Kirby, B.; Carr, D.B.; Buchanan, M.L.; Rutherford, T.; Thummel, K.E.; Fishbein, D.P.; Unadkat, J.D. Effects of pregnancy on CYP3A and P-glycoprotein activities as measured by disposition of midazolam and digoxin: A University of Washington specialized center of research study. Clin. Pharmacol. Ther. 2008, 84, 248–253. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Mendes, M.; Lui, G.; Zheng, Y.; Pressiat, C.; Hirt, D.; Valade, E.; Bouazza, N.; Foissac, F.; Blanche, S.; Treluyer, J.M.; et al. A physiologically-based pharmacokinetic model to predict human fetal exposure for a drug metabolized by several CYP450 pathways. Clin. Pharmacokinet. 2017, 56, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Heimbach, T.; Gollen, R.; Nanavati, C.; He, H. A simplified PBPK modeling approach for prediction of pharmacokinetics of four primarily renally excreted and CYP3A metabolized compounds during pregnancy. AAPS J. 2013, 15, 1012–1024. [Google Scholar] [CrossRef]
- Dallmann, A.; Ince, I.; Coboeken, K.; Eissing, T.; Hempel, G. A physiologically based pharmacokinetic model for pregnant women to predict the pharmacokinetics of drugs metabolized via several enzymatic pathways. Clin. Pharmacokinet. 2018, 57, 749–769. [Google Scholar] [CrossRef]
- Abduljalil, K.; Pansari, A.; Jamei, M. Prediction of maternal pharmacokinetics using physiologically based pharmacokinetic models: Assessing the impact of the longitudinal changes in the activity of CYP1A2, CYP2D6 and CYP3A4 enzymes during pregnancy. J. Pharmacokinet. Pharmacodyn. 2020, 47, 361–383. [Google Scholar] [CrossRef] [PubMed]
- Khatri, R.; Kulick, N.; Rementer, R.J.; Fallon, J.K.; Sykes, C.; Schauer, A.P.; Malinen, M.M.; Mosedale, M.; Watkins, P.B.; Kashuba, A.D.; et al. Pregnancy-related hormones increase nifedipine metabolism in human hepatocytes by inducing CYP3A4 expression. J. Pharm. Sci. 2021, 110, 412–421. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegaert, K.; Quinney, S.K.; Dallmann, A. Physiologically Based Pharmacokinetic Modeling in Pregnancy, during Lactation and in Neonates: Achievements, Challenges and Future Directions. Pharmaceutics 2024, 16, 500. https://doi.org/10.3390/pharmaceutics16040500
Allegaert K, Quinney SK, Dallmann A. Physiologically Based Pharmacokinetic Modeling in Pregnancy, during Lactation and in Neonates: Achievements, Challenges and Future Directions. Pharmaceutics. 2024; 16(4):500. https://doi.org/10.3390/pharmaceutics16040500
Chicago/Turabian StyleAllegaert, Karel, Sara K. Quinney, and André Dallmann. 2024. "Physiologically Based Pharmacokinetic Modeling in Pregnancy, during Lactation and in Neonates: Achievements, Challenges and Future Directions" Pharmaceutics 16, no. 4: 500. https://doi.org/10.3390/pharmaceutics16040500
APA StyleAllegaert, K., Quinney, S. K., & Dallmann, A. (2024). Physiologically Based Pharmacokinetic Modeling in Pregnancy, during Lactation and in Neonates: Achievements, Challenges and Future Directions. Pharmaceutics, 16(4), 500. https://doi.org/10.3390/pharmaceutics16040500