

Fundamentals and Applications of Focused Ultrasound-Assisted Cancer Immune Checkpoint Inhibition for Solid Tumors


Abstract
1. Introduction
2. Cancer from an Immune Point of View and Immune Check Point Inhibition
3. Immunological Barriers and TME Immune Profile
3.1. Metabolic Reprogramming: Adenosinergic Signaling
3.2. Immunosuppressive Cytokines: TGF-β and IL-10
3.3. Genomic Instability, Tumor Mutational Burden, and Tumor-Associated Antigens
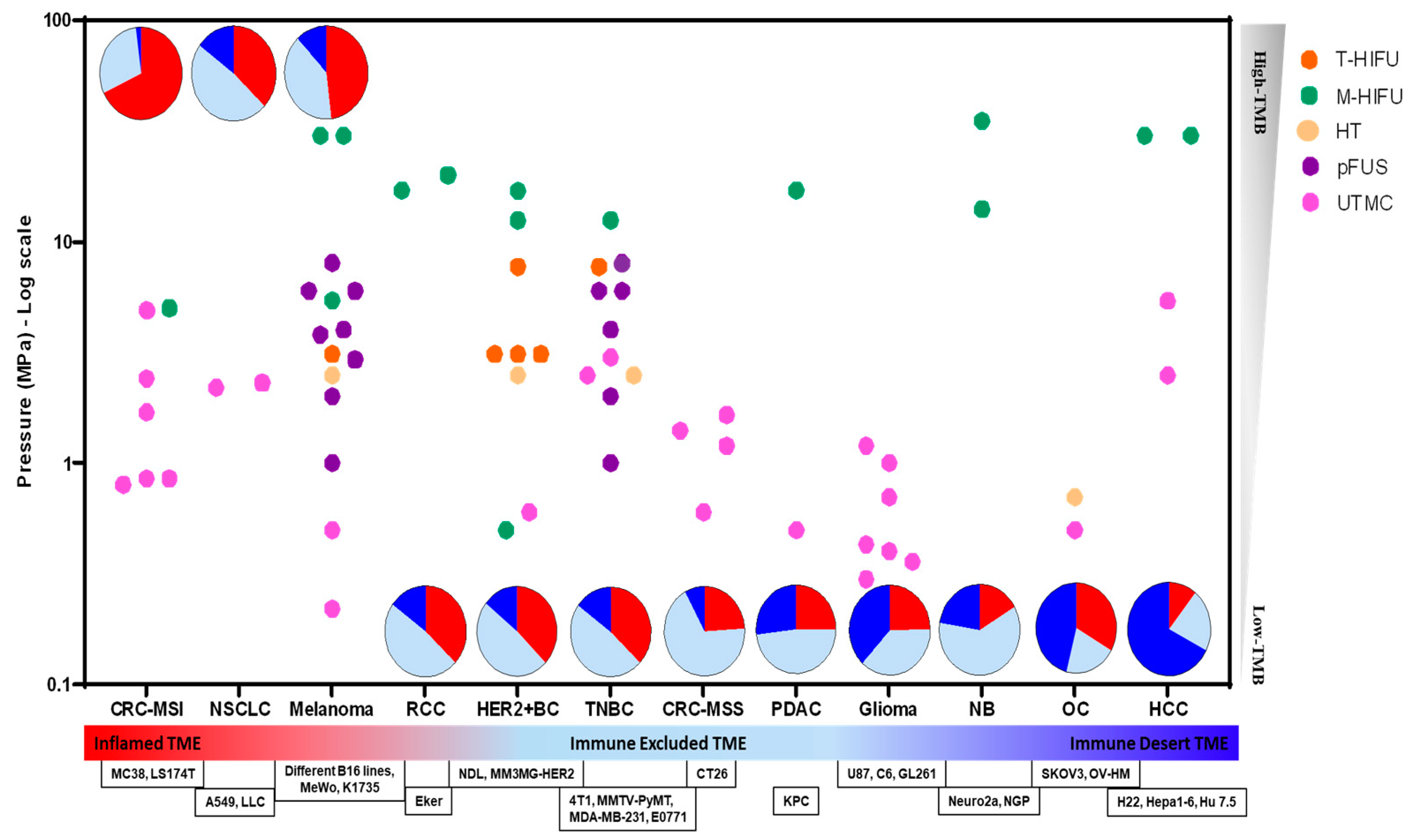
3.4. TME Immune Profiles and Resistance to ICI Therapy

4. Focused Ultrasound (FUS) Modalities for Cancer Therapy
4.1. Thermal Ablation HIFU (T-HIFU)
4.2. Mechanical Ablation HIFU (M-HIFU)
4.3. HIFU-Induced Hyperthermia (HT)
4.4. Pulsed FUS (pFUS)
4.5. Ultrasound-Targeted Microbubble Cavitation (UTMC)
5. Immune Profiles, FUS Bioeffects, and ICI

5.1. Ablative FUS + ICI
5.2. Non-Ablative FUS + ICI
6. Translational Challenges and Outlook
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sonpavde, G.P.; Grivas, P.; Lin, Y.; Hennessy, D.; Hunt, J.D. Immune-related adverse events with pd-1 versus pd-l1 inhibitors: A meta-analysis of 8730 patients from clinical trials. Future Oncol. 2021, 17, 2545–2558. [Google Scholar] [CrossRef] [PubMed]
- du Rusquec, P.; de Calbiac, O.; Robert, M.; Campone, M.; Frenel, J.S. Clinical utility of pembrolizumab in the management of advanced solid tumors: An evidence-based review on the emerging new data. Cancer Manag. Res. 2019, 11, 4297. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Paz-Ares, L.; Marreaud, S.; Dafni, U.; Oselin, K.; Havel, L.; Esteban, E.; Isla, D.; Martinez-Marti, A.; Faehling, M. Pembrolizumab versus placebo as adjuvant therapy for completely resected stage ib–iiia non-small-cell lung cancer (pearls/keynote-091): An interim analysis of a randomised, triple-blind, phase 3 trial. Lancet Oncol. 2022, 23, 1274–1286. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Harrington, K.; Greil, R.; Soulières, D.; Tahara, M.; De Castro, G.; Psyrri, A.; Rotllan, N.B.; Neupane, P.; Bratland, Å. Keynote-048: Phase III study of first-line pembrolizumab (P) for recurrent/metastatic head and neck squamous cell carcinoma (r/m hnscc). Ann. Oncol. 2018, 29, viii729. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Quinn, D.I.; Zhang, T.; Gurney, H.; Doshi, G.K.; Cobb, P.W.; Parnis, F.; Lee, J.-L.; Park, S.H.; Semenov, A. keynote-564: A phase 3, randomized, double blind, trial of pembrolizumab in the adjuvant treatment of renal cell carcinoma. J. Clin. Oncol. 2018, 36, TPS4599. [Google Scholar] [CrossRef]
- Joiner, J.B.; Pylayeva-Gupta, Y.; Dayton, P.A. Focused Ultrasound for Immunomodulation of the Tumor Microenvironment. J. Immunol. 2020, 205, 2327–2341. [Google Scholar] [CrossRef]
- Meng, Y.; Pople, C.B.; Lea-Banks, H.; Abrahao, A.; Davidson, B.; Suppiah, S.; Vecchio, L.M.; Samuel, N.; Mahmud, F.; Hynynen, K.; et al. Safety and efficacy of focused ultrasound induced blood-brain barrier opening, an integrative review of animal and human studies. J. Control Release 2019, 309, 25–36. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, X.Y.; Liu, Y.; Sankin, G.N.; Pua, E.C.; Morse, M.A.; Lyerly, H.K.; Clay, T.M.; Zhong, P. Investigation of HIFU-induced anti-tumor immunity in a murine tumor model. J. Transl. Med. 2007, 5, 34. [Google Scholar] [CrossRef]
- Snipstad, S.; Vikedal, K.; Maardalen, M.; Kurbatskaya, A.; Sulheim, E.; Davies, C.L. Ultrasound and microbubbles to beat barriers in tumors: Improving delivery of nanomedicine. Adv. Drug Deliv. Rev. 2021, 177, 113847. [Google Scholar] [CrossRef]
- Jung, O.; Thomas, A.; Burks, S.R.; Dustin, M.L.; Frank, J.A.; Ferrer, M.; Stride, E. Neuroinflammation associated with ultrasound-mediated permeabilization of the blood-brain barrier. Trends Neurosci. 2022, 45, 459–470. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Gubin, M.M.; Vesely, M.D. Cancer Immunoediting in the Era of Immuno-oncology. Clin. Cancer Res. 2022, 28, 3917–3928. [Google Scholar] [CrossRef] [PubMed]
- Varade, J.; Magadan, S.; Gonzalez-Fernandez, A. Human immunology and immunotherapy: Main achievements and challenges. Cell Mol. Immunol. 2021, 18, 805–828. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.M. A Nobel Prize-worthy pursuit: Cancer immunology and harnessing immunity to tumour neoantigens. Immunology 2018, 155, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase ii keynote-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef]
- Hui, R.; Gandhi, L.; Carcereny Costa, E.; Felip, E.; Ahn, M.-J.; Eder, J.P.; Balmanoukian, A.S.; Leighl, N.B.; Aggarwal, C.; Horn, L. Long-term OS for patients with advanced NSCLC enrolled in the keynote-001 study of pembrolizumab (pembro). J. Clin. Oncol. 2016, 34, 9026. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Powles, T.; Csőszi, T.; Özgüroğlu, M.; Matsubara, N.; Géczi, L.; Cheng, S.Y.; Fradet, Y.; Oudard, S.; Vulsteke, C.; Barrera, R.M. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (keynote-361): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 931–945. [Google Scholar] [CrossRef]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Hoyos Usta, E.; Yanez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. N. Engl. J. Med. 2021, 385, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Ardolino, L.; Joshua, A. Immune checkpoint inhibitors in malignancy. Aust. Prescr. 2019, 42, 62–67. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD 39 and CD 73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, C.; Schildberg, F.A.; Schurich, A.; Diehl, L.; Hegenbarth, S.I.; Endl, E.; Lacher, S.; Müller, C.E.; Frey, J.; Simeoni, L. Adenosine regulates cd8 t-cell priming by inhibition of membrane-proximal t-cell receptor signalling. Immunology 2009, 128, e728–e737. [Google Scholar] [CrossRef]
- Zhang, H.; Conrad, D.M.; Butler, J.J.; Zhao, C.; Blay, J.; Hoskin, D.W. Adenosine acts through a2 receptors to inhibit il-2-induced tyrosine phosphorylation of stat5 in T lymphocytes: Role of cyclic adenosine 3′, 5′-monophosphate and phosphatases. J. Immunol. 2004, 173, 932–944. [Google Scholar] [CrossRef]
- Sorrentino, C.; Hossain, F.; Rodriguez, P.C.; Sierra, R.A.; Pannuti, A.; Hatfield, S.; Osborne, B.A.; Minter, L.M.; Miele, L.; Morello, S. Adenosine A2A receptor stimulation inhibits tcr-induced Notch1 activation in cd8+ t-cells. Front. Immunol. 2019, 10, 162. [Google Scholar] [CrossRef]
- Romio, M.; Reinbeck, B.; Bongardt, S.; Hüls, S.; Burghoff, S.; Schrader, J. Extracellular purine metabolism and signaling of cd73-derived adenosine in murine Treg and Teff cells. Am. J. Physiol. Cell Physiol. 2011, 301, C530–C539. [Google Scholar] [CrossRef]
- Leone, R.D.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and act in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of cd4+ cd25+ foxp3+ regulatory T cells are under influence of the adenosine-a2a adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef]
- Zarek, P.E.; Huang, C.-T.; Lutz, E.R.; Kowalski, J.; Horton, M.R.; Linden, J.; Drake, C.G.; Powell, J.D. A2a receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 2008, 111, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of antitumor immunity by il-10 and tgf-β-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immun. 2006, 177, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Wrzesinski, S.H.; Wan, Y.Y.; Flavell, R.A. Transforming growth factor-β and the immune response: Implications for anticancer therapy. Clin. Cancer Res. 2007, 13, 5262–5270. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.M.; Mortimer, T.O.; O’Neill, K.L. Cytokines: Can Cancer Get the Message? Cancers 2022, 14, 2178. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, ra230–ra277. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A.; Blaser, K. Mechanisms of interleukin-10-mediated immune suppression. Immunology 2001, 103, 131–136. [Google Scholar] [CrossRef]
- Neven, B.; Mamessier, E.; Bruneau, J.; Kaltenbach, S.; Kotlarz, D.; Suarez, F.; Masliah-Planchon, J.; Billot, K.; Canioni, D.; Frange, P.; et al. A Mendelian predisposition to B-cell lymphoma caused by il-10r deficiency. Blood 2013, 122, 3713–3722. [Google Scholar] [CrossRef]
- Bai, X.; Yi, M.; Jiao, Y.; Chu, Q.; Wu, K. Blocking TGF-β signaling to enhance the efficacy of immune checkpoint inhibitor. OncoTargets Ther. 2019, 12, 9527–9538. [Google Scholar] [CrossRef]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA evolution: Old ideas, new approaches. Curr. Opin. Genet. Dev. 2018, 49, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Addeo, A.; Friedlaender, A.; Banna, G.L.; Weiss, G.J. TMB or not tmb as a biomarker: That is the question. Crit. Rev. Oncol. Hematol. 2021, 163, 103374. [Google Scholar] [CrossRef] [PubMed]
- Choucair, K.; Morand, S.; Stanbery, L.; Edelman, G.; Dworkin, L.; Nemunaitis, J. TMB: A promising immune-response biomarker, and potential spearhead in advancing targeted therapy trials. Cancer Gene Ther. 2020, 27, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Joshi, K.; de Massy, M.R.; Ismail, M.; Reading, J.L.; Uddin, I.; Woolston, A.; Hatipoglu, E.; Oakes, T.; Rosenthal, R.; Peacock, T.; et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat. Med. 2019, 25, 1549–1559. [Google Scholar] [CrossRef]
- Sugiura, A.; Rathmell, J.C. Metabolic Barriers to T Cell Function in Tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E. TGFβ attenuates tumour response to pd-l1 blockade by contributing to exclusion of T cells. Nature. Nat. 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Spranger, S.; Fuchs, E.; Lopez-Soto, A. WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 2019, 29, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, Z.; Izadifar, Z.; Chapman, D.; Babyn, P. An Introduction to High Intensity Focused Ultrasound: Systematic Review on Principles, Devices, and Clinical Applications. J. Clin. Med. 2020, 9, 460. [Google Scholar] [CrossRef]
- Shi, G.; Zhong, M.; Ye, F.; Zhang, X. Low-frequency HIFU induced cancer immunotherapy: Tempting challenges and potential opportunities. Cancer Biol. Med. 2019, 16, 714–728. [Google Scholar] [CrossRef] [PubMed]
- Bove, T.; Zawada, T.; Serup, J.; Jessen, A.; Poli, M. High-frequency (20-mhz) high-intensity focused ultrasound (hifu) system for dermal intervention: Preclinical evaluation in skin equivalents. Skin Res. Technol. 2019, 25, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Y.; Zou, J.Z.; Chen, Z.G.; Liu, S.; Jiao, J.; Wu, F. Acoustic Cavitation Enhances Focused Ultrasound Ablation with Phase-Shift Inorganic Perfluorohexane Nanoemulsions: An In Vitro Study Using a Clinical Device. Biomed. Res. Int. 2016, 2016, 7936902. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wong, C.O.; Cho, K.J.; van der Hoeven, D.; Liang, H.; Thakur, D.P.; Luo, J.; Babic, M.; Zinsmaier, K.E.; Zhu, M.X.; et al. SIGNAL TRANSDUCTION. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 2015, 349, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Feng, Y.; Ter Haar, G. Cavitation in Biomedicine; Springer: Berlin/Heidelberg, Germany, 2015; pp. 151–206. [Google Scholar]
- Kim, C.; Lim, M.; Woodworth, G.F.; Arvanitis, C.D. The roles of thermal and mechanical stress in focused ultrasound-mediated immunomodulation and immunotherapy for central nervous system tumors. J. Neurooncol. 2022, 157, 221–236. [Google Scholar] [CrossRef]
- Izadifar, Z.; Babyn, P.; Chapman, D. Mechanical and biological effects of ultrasound: A review of present knowledge. Ultrasound Med. Biol. 2017, 43, 1085–1104. [Google Scholar] [CrossRef]
- Riesberg, G.; Bigelow, T.A.; Stessman, D.J.; Spalding, M.H.; Yao, L.; Wang, T.; Xu, J. Flow rate and duty cycle effects in lysis of Chlamydomonas reinhardtii using high-energy pulsed focused ultrasound. J. Acoust. Soc. Am. 2014, 135, 3632–3638. [Google Scholar] [CrossRef]
- Holt, R.G.; Roy, R.A. Measurements of bubble-enhanced heating from focused, mhz-frequency ultrasound in a tissue-mimicking material. Ultrasound Med. Biol. 2001, 27, 1399–1412. [Google Scholar] [CrossRef]
- Xu, Z.; Hall, T.L.; Vlaisavljevich, E.; Lee, F.T., Jr. Histotripsy: The first noninvasive, non-ionizing, non-thermal ablation technique based on ultrasound. Int. J. Hyperth. 2021, 38, 561–575. [Google Scholar] [CrossRef]
- Guillemin, P.C.; Gui, L.; Lorton, O.; Zilli, T.; Crowe, L.A.; Desgranges, S.; Montet, X.; Terraz, S.; Miralbell, R.; Salomir, R. Mild hyperthermia by mr-guided focused ultrasound in an ex vivo model of osteolytic bone tumour: Optimization of the spatio-temporal control of the delivered temperature. J. Transl. Med. 2019, 17, 350. [Google Scholar] [CrossRef] [PubMed]
- Speed, C. Therapeutic ultrasound in soft tissue lesions. Rheumatology 2001, 40, 1331–1336. [Google Scholar] [CrossRef]
- Partanen, A.; Yarmolenko, P.S.; Viitala, A.; Appanaboyina, S.; Haemmerich, D.; Ranjan, A.; Jacobs, G.; Woods, D.; Enholm, J.; Wood, B.J. Mild hyperthermia with magnetic resonance-guided high-intensity focused ultrasound for applications in drug delivery. Int. J. Hyperth. 2012, 28, 320–336. [Google Scholar] [CrossRef] [PubMed]
- Frazier, N.; Payne, A.; de Bever, J.; Dillon, C.; Panda, A.; Subrahmanyam, N.; Ghandehari, H. High intensity focused ultrasound hyperthermia for enhanced macromolecular delivery. J. Control Release 2016, 241, 186–193. [Google Scholar] [CrossRef]
- Kheirolomoom, A.; Silvestrini, M.T.; Ingham, E.S.; Mahakian, L.M.; Tam, S.M.; Tumbale, S.K.; Foiret, J.; Hubbard, N.E.; Borowsky, A.D.; Ferrara, K.W. Combining activatable nanodelivery with immunotherapy in a murine breast cancer model. J. Control Release 2019, 303, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Aydin, O.; Chandran, P.; Lorsung, R.R.; Cohen, G.; Burks, S.R.; Frank, J.A. The Proteomic Effects of Pulsed Focused Ultrasound on Tumor Microenvironments of Murine Melanoma and Breast Cancer Models. Ultrasound Med. Biol. 2019, 45, 3232–3245. [Google Scholar] [CrossRef]
- Cohen, G.; Chandran, P.; Lorsung, R.M.; Aydin, O.; Tomlinson, L.E.; Rosenblatt, R.B.; Burks, S.R.; Frank, J.A. Pulsed-Focused Ultrasound Slows B16 Melanoma and 4T1 Breast Tumor Growth through Differential Tumor Microenvironmental Changes. Cancers 2021, 13, 1546. [Google Scholar] [CrossRef] [PubMed]
- Mohammadabadi, A.; Huynh, R.N.; Wadajkar, A.S.; Lapidus, R.G.; Kim, A.J.; Raub, C.B.; Frenkel, V. Pulsed focused ultrasound lowers interstitial fluid pressure and increases nanoparticle delivery and penetration in head and neck squamous cell carcinoma xenograft tumors. Phys. Med. Biol. 2020, 65, 125017. [Google Scholar] [CrossRef] [PubMed]
- Michon, S.; Rodier, F.; Yu, F.T.H. Targeted Anti-Cancer Provascular Therapy Using Ultrasound, Microbubbles, and Nitrite to Increase Radiotherapy Efficacy. Bioconjug. Chem. 2022, 33, 1093–1105. [Google Scholar] [CrossRef]
- Elhelf, I.A.S.; Albahar, H.; Shah, U.; Oto, A.; Cressman, E.; Almekkawy, M. High intensity focused ultrasound: The fundamentals, clinical applications and research trends. Diagn. Interv. Imaging 2018, 99, 349–359. [Google Scholar] [CrossRef]
- Ho, Y.J.; Li, J.P.; Fan, C.H.; Liu, H.L.; Yeh, C.K. Ultrasound in tumor immunotherapy: Current status and future developments. J. Control Release 2020, 323, 12–23. [Google Scholar] [CrossRef]
- Xia, J.Z.; Xie, F.L.; Ran, L.F.; Xie, X.P.; Fan, Y.M.; Wu, F. High-intensity focused ultrasound tumor ablation activates autologous tumor-specific cytotoxic T lymphocytes. Ultrasound Med. Biol. 2012, 38, 1363–1371. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, Y.; Feng, J.; Wu, F. Dendritic cells loaded with ultrasound-ablated tumour induce in vivo specific antitumour immune responses. Ultrasound Med. Biol. 2010, 36, 441–448. [Google Scholar] [CrossRef]
- Fite, B.Z.; Wang, J.; Kare, A.J.; Ilovitsh, A.; Chavez, M.; Ilovitsh, T.; Zhang, N.; Chen, W.; Robinson, E.; Zhang, H.; et al. Immune modulation resulting from mr-guided high intensity focused ultrasound in a model of murine breast cancer. Sci. Rep. 2021, 11, 927. [Google Scholar] [CrossRef] [PubMed]
- Silvestrini, M.T.; Ingham, E.S.; Mahakian, L.M.; Kheirolomoom, A.; Liu, Y.; Fite, B.Z.; Tam, S.M.; Tucci, S.T.; Watson, K.D.; Wong, A.W. Priming is key to effective incorporation of image-guided thermal ablation into immunotherapy protocols. JCI Insight 2017, 2, e90521. [Google Scholar] [CrossRef] [PubMed]
- Lundt, J.E.; Allen, S.P.; Shi, J.; Hall, T.L.; Cain, C.A.; Xu, Z. Non-invasive, Rapid Ablation of Tissue Volume Using Histotripsy. Ultrasound Med. Biol. 2017, 43, 2834–2847. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, Z.; Babyn, P.; Chapman, D. Ultrasound cavitation/microbubble detection and medical applications. J. Med. Biol. Eng. 2019, 39, 259–276. [Google Scholar] [CrossRef]
- Khokhlova, V.A.; Fowlkes, J.B.; Roberts, W.W.; Schade, G.R.; Xu, Z.; Khokhlova, T.D.; Hall, T.L.; Maxwell, A.D.; Wang, Y.N.; Cain, C.A. Histotripsy methods in mechanical disintegration of tissue: Towards clinical applications. Int. J. Hyperth. 2015, 31, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Vlaisavljevich, E.; Kim, Y.; Owens, G.; Roberts, W.; Cain, C.; Xu, Z. Effects of tissue mechanical properties on susceptibility to histotripsy-induced tissue damage. Phys. Med. Biol. 2014, 59, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Tang, J.; Yang, J.; Zhu, B.; Wang, X.; Luo, Y.; Yang, H.; Jang, F.; Zou, J.; Liu, Z.; et al. Tumor perfusion enhancement by ultrasound stimulated microbubbles potentiates PD-L1 blockade of MC38 colon cancer in mice. Cancer Lett. 2021, 498, 121–129. [Google Scholar] [CrossRef]
- Korbelik, M.; Banath, J.; Saw, K.M.; Zhang, W.; Čiplys, E. Calreticulin as cancer treatment adjuvant: Combination with photodynamic therapy and photodynamic therapy-generated vaccines. Front. Oncol. 2015, 5, 15. [Google Scholar] [CrossRef]
- Qu, S.; Worlikar, T.; Felsted, A.E.; Ganguly, A.; Beems, M.V.; Hubbard, R.; Pepple, A.L.; Kevelin, A.A.; Garavaglia, H.; Dib, J. Non-thermal histotripsy tumor ablation promotes abscopal immune responses that enhance cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000200. [Google Scholar] [CrossRef]
- Pahk, K.J.; Shin, C.H.; Bae, I.Y.; Yang, Y.; Kim, S.H.; Pahk, K.; Kim, H.; Oh, S.J. Boiling Histotripsy-induced Partial Mechanical Ablation Modulates Tumour Microenvironment by Promoting Immunogenic Cell Death of Cancers. Sci. Rep. 2019, 9, 9050. [Google Scholar] [CrossRef]
- Pisetsky, D. Cell death in the pathogenesis of immune-mediated diseases: The role of HMGB1 and DAMP-PAMP complexes. Swiss Med. Wkly. 2011, 141, w13256. [Google Scholar] [CrossRef]
- Iwanicki, I.; Wu, L.L.; Flores-Guzman, F.; Sundland, R.; Viza-Gomes, P.; Nordgren, R.; Centner, C.S.; Kandel, J.J.; Applebaum, M.A.; Bader, K.B.; et al. Histotripsy induces apoptosis and reduces hypoxia in a neuroblastoma xenograft model. Int. J. Hyperth. 2023, 40, 2222941. [Google Scholar] [CrossRef] [PubMed]
- Eranki, A.; Srinivasan, P.; Ries, M.; Kim, A.; Lazarski, C.A.; Rossi, C.T.; Khokhlova, T.D.; Wilson, E.; Knoblach, S.M.; Sharma, K.V. High-Intensity Focused Ultrasound (HIFU) Triggers Immune Sensitization of Refractory Murine Neuroblastoma to Checkpoint Inhibitor TherapyHIFU with Immunotherapy Cure Refractory Murine Neuroblastoma. Clin. Cancer Res. 2020, 26, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Schade, G.R.; Wang, Y.N.; D’Andrea, S.; Hwang, J.H.; Liles, W.C.; Khokhlova, T.D. Boiling Histotripsy Ablation of Renal Cell Carcinoma in the Eker Rat Promotes a Systemic Inflammatory Response. Ultrasound Med. Biol. 2019, 45, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Pepple, A.L.; Guy, J.L.; McGinnis, R.; Felsted, A.E.; Song, B.; Hubbard, R.; Worlikar, T.; Garavaglia, H.; Dib, J.; Chao, H.; et al. Spatiotemporal local and abscopal cell death and immune responses to histotripsy focused ultrasound tumor ablation. Front. Immunol. 2023, 14, 1012799. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, M.; Eikelenboom, D.; den Brok, M.H.; Veltien, A.; Wassink, M.; Wesseling, P.; Dumont, E.; Futterer, J.J.; Adema, G.J.; Heerschap, A. In vivo MR guided boiling histotripsy in a mouse tumor model evaluated by MRI and histopathology. NMR Biomed. 2016, 29, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Skandalakis, G.P.; Rivera, D.R.; Rizea, C.D.; Bouras, A.; Jesu Raj, J.G.; Bozec, D.; Hadjipanayis, C.G. Hyperthermia treatment advances for brain tumors. Int. J. Hyperth. 2020, 37, 3–19. [Google Scholar] [CrossRef]
- Suzuki, R.; Namai, E.; Oda, Y.; Nishiie, N.; Otake, S.; Koshima, R.; Hirata, K.; Taira, Y.; Utoguchi, N.; Negishi, Y.; et al. Cancer gene therapy by il-12 gene delivery using liposomal bubbles and tumoral ultrasound exposure. J. Control Release 2010, 142, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Yang, K.J.; Wu, J.C.; Chang, K.J.; Wang, S.M. Effects of hyperthermia on the cytoskeleton and focal adhesion proteins in a human thyroid carcinoma cell line. J. Cell Biochem. 1999, 75, 327–337. [Google Scholar] [CrossRef]
- Oei, A.L.; Vriend, L.E.; Crezee, J.; Franken, N.A.; Krawczyk, P.M. Effects of hyperthermia on DNA repair pathways: One treatment to inhibit them all. Radiat. Oncol. 2015, 10, 165. [Google Scholar] [CrossRef]
- Ostberg, J.R.; Repasky, E.A. Emerging evidence indicates that physiologically relevant thermal stress regulates dendritic cell function. Cancer Immunol. Immunother. 2006, 55, 292–298. [Google Scholar] [CrossRef]
- Gouarderes, S.; Mingotaud, A.-F.; Vicendo, P.; Gibot, L. Vascular and extracellular matrix remodeling by physical approaches to improve drug delivery at the tumor site. Expert. Opin. Drug Deliv. 2020, 17, 1703–1726. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, Q.; Dukhovlinova, E.; Chen, G.; Ahn, S.; Wang, C.; Ogunnaike, E.A.; Ligler, F.S.; Dotti, G.; Gu, Z. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of car t Cells. Adv. Mater. 2019, 31, e1900192. [Google Scholar] [CrossRef]
- Chang, M.; Hou, Z.; Wang, M.; Li, C.; Lin, J. Recent Advances in Hyperthermia Therapy-Based Synergistic Immunotherapy. Adv. Mater. 2021, 33, e2004788. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Suzuki, Y.; Kaminuma, T.; Yoshimoto, Y.; Oike, T.; Okonogi, N.; Sato, H.; Tamaki, T.; Noda, S.-E.; Mimura, K. Tumor-specific CD8-positive T cell-mediated antitumor immunity is implicated in the antitumor effect of local hyperthermia. Int. J. Hyperth. 2018, 35, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Dayanc, B.E.; Beachy, S.H.; Ostberg, J.R.; Repasky, E.A. Dissecting the role of hyperthermia in natural killer cell mediated anti-tumor responses. Int. J. Hyperth. 2008, 24, 41–56. [Google Scholar] [CrossRef]
- Chen, Q.; Appenheimer, M.M.; Muhitch, J.B.; Fisher, D.T.; Clancy, K.A.; Miecznikowski, J.C.; Wang, W.C.; Evans, S.S. Thermal facilitation of lymphocyte trafficking involves temporal induction of intravascular ICAM-1. Microcirculation 2009, 16, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the thermal regulation of immunity: The immune system feels the heat. Nat. Rev. Immunol. 2015, 15, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Sheybani, N.D.; Witter, A.R.; Thim, E.A.; Yagita, H.; Bullock, T.N.J.; Price, R.J. Combination of thermally ablative focused ultrasound with gemcitabine controls breast cancer via adaptive immunity. J. Immunother. Cancer 2020, 8, e001008. [Google Scholar] [CrossRef]
- Han, X.; Wang, R.; Xu, J.; Chen, Q.; Liang, C.; Chen, J.; Zhao, J.; Chu, J.; Fan, Q.; Archibong, E.; et al. In situ thermal ablation of tumors in combination with nano-adjuvant and immune checkpoint blockade to inhibit cancer metastasis and recurrence. Biomaterials 2019, 224, 119490. [Google Scholar] [CrossRef]
- Burks, S.R.; Ziadloo, A.; Hancock, H.A.; Chaudhry, A.; Dean, D.D.; Lewis, B.K.; Frenkel, V.; Frank, J.A. Investigation of cellular and molecular responses to pulsed focused ultrasound in a mouse model. PLoS ONE 2011, 6, e24730. [Google Scholar] [CrossRef]
- Li, T.; Wang, Y.N.; Khokhlova, T.D.; D’Andrea, S.; Starr, F.; Chen, H.; McCune, J.S.; Risler, L.J.; Mashadi-Hossein, A.; Hingorani, S.R.; et al. Pulsed High-Intensity Focused Ultrasound Enhances Delivery of Doxorubicin in a Preclinical Model of Pancreatic Cancer. Cancer Res. 2015, 75, 3738–3746. [Google Scholar] [CrossRef]
- Lee, S.; Han, H.; Koo, H.; Na, J.H.; Yoon, H.Y.; Lee, K.E.; Lee, H.; Kim, H.; Kwon, I.C.; Kim, K. Extracellular matrix remodeling in vivo for enhancing tumor-targeting efficiency of nanoparticle drug carriers using the pulsed high intensity focused ultrasound. J. Control Release 2017, 263, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, F.; Shigemura, K.; Maeda, K.; Hiraoka, A.; Maeshige, N.; Ooya, T.; Sung, S.Y.; Yang, Y.M.; Fujisawa, M. Combined Treatment with Ultrasound and Immune Checkpoint Inhibitors for Prostate Cancer. J. Clin. Med. 2022, 11, 2448. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Quinn, T.J.; Scandiuzzi, L.; Basu, I.; Partanen, A.; Tome, W.A.; Macian, F.; Guha, C. Low-Intensity Focused Ultrasound Induces Reversal of Tumor-Induced t Cell Tolerance and Prevents Immune Escape. J. Immunol. 2016, 196, 1964–1976. [Google Scholar] [CrossRef]
- Sirsi, S.R.; Borden, M.A. Advances in ultrasound mediated gene therapy using microbubble contrast agents. Theranostics 2012, 2, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Frinking, P.; Segers, T.; Luan, Y.; Tranquart, F. Three Decades of Ultrasound Contrast Agents: A Review of the Past, Present and Future Improvements. Ultrasound Med. Biol. 2020, 46, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.M.; Abou-Elkacem, L.; Lee, T.; Dahl, J.; Lutz, A.M. Ultrasound and microbubble mediated therapeutic delivery: Underlying mechanisms and future outlook. J. Control Release 2020, 326, 75–90. [Google Scholar] [CrossRef]
- Tran, T.A.; Le Guennec, J.Y.; Bougnoux, P.; Tranquart, F.; Bouakaz, A. Characterization of cell membrane response to ultrasound activated microbubbles. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2008, 55, 43–49. [Google Scholar] [CrossRef]
- Kooiman, K.; van der Steen, A.F.; de Jong, N. Role of intracellular calcium and reactive oxygen species in microbubble-mediated alterations of endothelial layer permeability. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2013, 60, 1811–1815. [Google Scholar] [CrossRef]
- Beekers, I.; Mastik, F.; Beurskens, R.; Tang, P.Y.; Vegter, M.; van der Steen, A.F.W.; de Jong, N.; Verweij, M.D.; Kooiman, K. High-Resolution Imaging of Intracellular Calcium Fluctuations Caused by Oscillating Microbubbles. Ultrasound Med. Biol. 2020, 46, 2017–2029. [Google Scholar] [CrossRef]
- Belcik, J.T.; Davidson, B.P.; Xie, A.; Wu, M.D.; Yadava, M.; Qi, Y.; Liang, S.; Chon, C.R.; Ammi, A.Y.; Field, J. Augmentation of muscle blood flow by ultrasound cavitation is mediated by ATP and purinergic signaling. Circulation 2017, 135, 1240–1252. [Google Scholar] [CrossRef]
- Grygorczyk, R.; Boudreault, F.; Ponomarchuk, O.; Tan, J.J.; Furuya, K.; Goldgewicht, J.; Kenfack, F.D.; Yu, F. Lytic Release of Cellular atp: Physiological Relevance and Therapeutic Applications. Life 2021, 11, 700. [Google Scholar] [CrossRef]
- Haugse, R.; Langer, A.; Murvold, E.T.; Costea, D.E.; Gjertsen, B.T.; Gilja, O.H.; Kotopoulis, S.; Ruiz de Garibay, G.; McCormack, E. Low-intensity sonoporation-induced intracellular signalling of pancreatic cancer cells, fibroblasts and endothelial cells. Pharmaceutics 2020, 12, 1058. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, B.; Ding, H.; Shi, H.; Liu, J.; Sun, H. Low-intensity pulsed ultrasound in combination with SonoVue induces cytotoxicity of human renal glomerular endothelial cells via repression of the erk1/2 signaling pathway. Ren. Fail. 2018, 40, 458–465. [Google Scholar] [CrossRef]
- Saha, S.; Bhanja, P.; Partanen, A.; Zhang, W.; Liu, L.; Tomé, W.; Guha, C. Low intensity focused ultrasound (lofu) modulates unfolded protein response and sensitizes prostate cancer to 17aag. Oncoscience 2014, 1, 434. [Google Scholar] [CrossRef] [PubMed]
- Amate, M.; Goldgewicht, J.; Sellamuthu, B.; Stagg, J.; Yu, F.T.H. The effect of ultrasound pulse length on microbubble cavitation induced antibody accumulation and distribution in a mouse model of breast cancer. Nanotheranostics 2020, 4, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, V. Ultrasound mediated delivery of drugs and genes to solid tumors. Adv. Drug Deliv. Rev. 2008, 60, 1193–1208. [Google Scholar] [CrossRef]
- Yang, C.; Du, M.; Yan, F.; Chen, Z. Focused ultrasound improves NK-92MI cells infiltration into tumors. Front. Pharmacol. 2019, 10, 326. [Google Scholar] [CrossRef]
- Qin, J.; Wang, T.-Y.; Willmann, J.K. Sonoporation: Applications for Cancer Therapy. In Therapeutic Ultrasound; Escoffre, J.-M., Bouakaz, A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 263–291. [Google Scholar]
- Beekers, I.; Vegter, M.; Lattwein, K.R.; Mastik, F.; Beurskens, R.; van der Steen, A.F.; de Jong, N.; Verweij, M.D.; Kooiman, K. Opening of endothelial cell–cell contacts due to sonoporation. J. Control Release 2020, 322, 426–438. [Google Scholar] [CrossRef]
- Heath, C.H.; Sorace, A.; Knowles, J.; Rosenthal, E.; Hoyt, K. Microbubble therapy enhances anti-tumor properties of cisplatin and cetuximab in vitro and in vivo. Otolaryngol. Head. Neck Surg. 2012, 146, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Zderic, V.; Clark, J.I.; Vaezy, S. Drug delivery into the eye with the use of ultrasound. J. Ultrasound Med. 2004, 23, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Schoellhammer, C.M.; Schroeder, A.; Maa, R.; Lauwers, G.Y.; Swiston, A.; Zervas, M.; Barman, R.; DiCiccio, A.M.; Brugge, W.R.; Anderson, D.G.; et al. Ultrasound-mediated gastrointestinal drug delivery. Sci. Transl. Med. 2015, 7, 310ra168. [Google Scholar] [CrossRef] [PubMed]
- Azagury, A.; Khoury, L.; Enden, G.; Kost, J. Ultrasound mediated transdermal drug delivery. Adv. Drug Deliv. Rev. 2014, 72, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Hsieh, H.Y.; Huang, C.Y.; Lin, C.Y.; Wei, K.C.; Liu, H.L. Focused ultrasound-induced blood-brain barrier opening to enhance interleukin-12 delivery for brain tumor immunotherapy: A preclinical feasibility study. J. Transl. Med. 2015, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Janowicz, P.W.; Leinenga, G.; Gotz, J.; Nisbet, R.M. Ultrasound-mediated blood-brain barrier opening enhances delivery of therapeutically relevant formats of a tau-specific antibody. Sci. Rep. 2019, 9, 9255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sierra, C.; Kwon, N.; Karakatsani, M.E.; Jackson-Lewis, V.R.; Przedborski, S.; Konofagou, E. Focused-Ultrasound Mediated Anti-Alpha-Synuclein Antibody Delivery for the Treatment of Parkinson’s Disease. In Proceedings of the 2018 IEEE International Ultrasonics Symposium, Kobe, Japan, 22–25 October 2018; pp. 1–4. [Google Scholar]
- Kovacs, Z.I.; Kim, S.; Jikaria, N.; Qureshi, F.; Milo, B.; Lewis, B.K.; Bresler, M.; Burks, S.R.; Frank, J.A. Disrupting the blood-brain barrier by focused ultrasound induces sterile inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E75–E84. [Google Scholar] [CrossRef]
- McMahon, D.; Hynynen, K. Acute inflammatory response following increased blood-brain barrier permeability induced by focused ultrasound is dependent on microbubble dose. Theranostics 2017, 7, 3989. [Google Scholar] [CrossRef]
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.Y.; Fang, D.; Latha, K.; et al. Opening of the Blood-Brain Barrier Using Low-Intensity Pulsed Ultrasound Enhances Responses to Immunotherapy in Preclinical Glioma Models. Clin. Cancer Res. 2021, 27, 4325–4337. [Google Scholar] [CrossRef] [PubMed]
- Curley, C.T.; Mead, B.P.; Negron, K.; Kim, N.; Garrison, W.J.; Miller, G.W.; Kingsmore, K.M.; Thim, E.A.; Song, J.; Munson, J.M.; et al. Augmentation of brain tumor interstitial flow via focused ultrasound promotes brain-penetrating nanoparticle dispersion and transfection. Sci. Adv. 2020, 6, eaay1344. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Reilly, R.M.; Pezo, R.C.; Trudeau, M.; Sahgal, A.; Singnurkar, A.; Perry, J.; Myrehaug, S.; Pople, C.B.; Davidson, B.; et al. MR-guided focused ultrasound enhances delivery of trastuzumab to her2-positive brain metastases. Sci. Transl. Med. 2021, 13, eabj4011. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Liu, J.; Liao, L.; Sun, J.; Jin, W.; Shu, X. Ultrasound Combined with Microbubbles Increase the Delivery of Doxorubicin by Reducing the Interstitial Fluid Pressure. Ultrasound Q. 2019, 35, 103–109. [Google Scholar] [CrossRef]
- Zhang, Q.; Jin, H.; Chen, L.; Chen, Q.; He, Y.; Yang, Y.; Ma, S.; Xiao, S.; Xi, F.; Luo, Q.; et al. Effect of Ultrasound Combined With Microbubble Therapy on Interstitial Fluid Pressure and VX2 Tumor Structure in Rabbit. Front. Pharmacol. 2019, 10, 716. [Google Scholar] [CrossRef]
- Eisenbrey, J.R.; Shraim, R.; Liu, J.-B.; Li, J.; Stanczak, M.; Oeffinger, B.; Leeper, D.B.; Keith, S.W.; Jablonowski, L.J.; Forsberg, F. Sensitization of hypoxic tumors to radiation therapy using ultrasound-sensitive oxygen microbubbles. Int. Res. J. Biotechnol. 2018, 101, 88–96. [Google Scholar] [CrossRef]
- Ho, Y.J.; Chu, S.W.; Liao, E.C.; Fan, C.H.; Chan, H.L.; Wei, K.C.; Yeh, C.K. Normalization of Tumor Vasculature by Oxygen Microbubbles with Ultrasound. Theranostics 2019, 9, 7370–7383. [Google Scholar] [CrossRef]
- Chin, C.T.; Raju, B.I.; Shevchenko, T.; Klibanov, A.L. Control and Reversal of Tumor Growth by Ultrasound Activated Microbubbles. In Proceedings of the 2009 IEEE International Ultrasonics. Symposium, Rome, Italy, 20–23 September 2009; pp. 77–80. [Google Scholar]
- Burke, C.W.; Klibanov, A.L.; Sheehan, J.P.; Price, R.J. Inhibition of glioma growth by microbubble activation in a subcutaneous model using low duty cycle ultrasound without significant heating. J. Neurosurg. 2011, 114, 1654–1661. [Google Scholar] [CrossRef]
- Daecher, A.; Stanczak, M.; Liu, J.B.; Zhang, J.; Du, S.; Forsberg, F.; Leeper, D.B.; Eisenbrey, J.R. Localized microbubble cavitation-based antivascular therapy for improving HCC treatment response to radiotherapy. Cancer Lett. 2017, 411, 100–105. [Google Scholar] [CrossRef]
- Goertz, D.E.; Karshafian, R.; Hynynen, K. Investigating the Effects of Pulsed Low Intensity Ultrasound and Microbubbles in Mouse Tumors. In Proceedings of the 2009 IEEE International Ultrasonics. Symposium, Rome, Italy, 20–23 September 2009; pp. 89–92. [Google Scholar]
- El Kaffas, A.; Gangeh, M.J.; Farhat, G.; Tran, W.T.; Hashim, A.; Giles, A.; Czarnota, G.J. Tumour Vascular Shutdown and Cell Death Following Ultrasound-Microbubble Enhanced Radiation Therapy. Theranostics 2018, 8, 314–327. [Google Scholar] [CrossRef]
- Skalina, K.A.; Singh, S.; Chavez, C.G.; Macian, F.; Guha, C. Low intensity focused ultrasound (lofu)-mediated acoustic immune priming and ablative radiation therapy for in situ tumor vaccines. Sci. Rep. 2019, 9, 15516. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.J.; Gade, T.; Soulen, M.C.; Pickup, S.; Sehgal, C.M. Antivascular ultrasound therapy: Magnetic resonance imaging validation and activation of the immune response in murine melanoma. J. Ultrasound Med. 2015, 34, 275–287. [Google Scholar] [CrossRef]
- Jahangiri, S.; Stagg, J.; Yu, F. UTMC Effect on Cancer Cell Apoptosis, Proliferation, and Vascular Inflammation in Wild Type and CD39 Knock Out Mice Model of MC38 Colon Cancer. In Proceedings of the 2023 IEEE International Ultrasonics. Symposium, Montreal, QC, Canada, 3–8 September 2023; pp. 1–2. [Google Scholar]
- Lin, L.; Du, Y.; Hao, J.; Wu, R.; Du, L. UTMD inhibits pancreatic cancer growth and metastasis by inducing macrophage polarization and vessel normalization. Biomed. Pharmacother. 2023, 160, 114322. [Google Scholar] [CrossRef] [PubMed]
- Joiner, J.B.; Kren, N.P.; Durham, P.G.; McRee, A.J.; Dayton, P.A.; Pylayeva-Gupta, Y. Low-Intensity Focused Ultrasound Produces Immune Response in Pancreatic Cancer. Ultrasound Med. Biol. 2022, 48, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Hsieh, H.Y.; Lu, L.A.; Kang, C.W.; Wu, M.F.; Lin, C.Y. Low-pressure pulsed focused ultrasound with microbubbles promotes an anticancer immunological response. J. Transl. Med. 2012, 10, 221. [Google Scholar] [CrossRef]
- Curley, C.T.; Stevens, A.D.; Mathew, A.S.; Stasiak, K.; Garrison, W.J.; Miller, G.W.; Sheybani, N.D.; Engelhard, V.H.; Bullock, T.N.J.; Price, R.J. Immunomodulation of intracranial melanoma in response to blood-tumor barrier opening with focused ultrasound. Theranostics 2020, 10, 8821–8833. [Google Scholar] [CrossRef]
- Escors, D. Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J. Sci. 2014, 2014, 734515. [Google Scholar] [CrossRef]
- Dai, Q.; Wilhelm, S.; Ding, D.; Syed, A.M.; Sindhwani, S.; Zhang, Y.; Chen, Y.Y.; MacMillan, P.; Chan, W.C.W. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano 2018, 12, 8423–8435. [Google Scholar] [CrossRef]
- Deprez, J.; Lajoinie, G.; Engelen, Y.; De Smedt, S.C.; Lentacker, I. Opening doors with ultrasound and microbubbles: Beating biological barriers to promote drug delivery. Adv. Drug Deliv. Rev. 2021, 172, 9–36. [Google Scholar] [CrossRef]
- Bulner, S.; Prodeus, A.; Gariepy, J.; Hynynen, K.; Goertz, D.E. Enhancing Checkpoint Inhibitor Therapy with Ultrasound Stimulated Microbubbles. Ultrasound Med. Biol. 2019, 45, 500–512. [Google Scholar] [CrossRef]
- Vos, H.; Lambein, K.; Richard, F.; Marien, B.; Nevelsteen, I.; Punie, K.; Wildiers, H.; Berben, L.; Laenen, A.; Floris, G.; et al. Comparison of the tumor immune microenvironment of primary hormone receptor-negative HER2-positive and triple negative breast cancer. NPJ Breast Cancer 2021, 7, 128. [Google Scholar] [CrossRef]
- Voorwerk, L.; Sanders, J.; Keusters, M.S.; Balduzzi, S.; Cornelissen, S.; Duijst, M.; Lips, E.H.; Sonke, G.S.; Linn, S.C.; Horlings, H.M.; et al. Immune landscape of breast tumors with low and intermediate estrogen receptor expression. NPJ Breast Cancer 2023, 9, 39. [Google Scholar] [CrossRef]
- Masih, K.E.; Wei, J.S.; Milewski, D.; Khan, J. Exploring and Targeting the Tumor Immune Microenvironment of Neuroblastoma. J. Cell. Immunol. 2021, 3, 305–316. [Google Scholar]
- Zhang, C.; Guo, L.; Su, Z.; Luo, N.; Tan, Y.; Xu, P.; Ye, L.; Tong, S.; Liu, H.; Li, X.; et al. Tumor Immune Microenvironment Landscape in Glioma Identifies a Prognostic and Immunotherapeutic Signature. Front. Cell Dev. Biol. 2021, 9, 717601. [Google Scholar] [CrossRef]
- Yang, B.; Li, X.; Zhang, W.; Fan, J.; Zhou, Y.; Li, W.; Yin, J.; Yang, X.; Guo, E.; Li, X.; et al. Spatial heterogeneity of infiltrating T cells in high-grade serous ovarian cancer revealed by multi-omics analysis. Cell Rep. Med. 2022, 3, 100856. [Google Scholar] [CrossRef] [PubMed]
- Karpinski, P.; Rossowska, J.; Sasiadek, M.M. Immunological landscape of consensus clusters in colorectal cancer. Oncotarget 2017, 8, 105299–105311. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Chavez, M.; Silvestrini, M.T.; Ingham, E.S.; Fite, B.Z.; Mahakian, L.M.; Tam, S.M.; Ilovitsh, A.; Monjazeb, A.M.; Murphy, W.J.; Hubbard, N.E.; et al. Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics 2018, 8, 3611–3628. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Nagata, H.; Crosby, E.J.; Inoue, Y.; Kaneko, K.; Liu, C.X.; Yang, X.; Wang, T.; Acharya, C.R.; Agarwal, P.; et al. Combination of ultrasound-based mechanical disruption of tumor with immune checkpoint blockade modifies tumor microenvironment and augments systemic antitumor immunity. J. Immunother. Cancer 2022, 10, e003717. [Google Scholar] [CrossRef] [PubMed]
- Goertz, D.; Cruz, W.; Bulner, S.; Wright, A.; Kerbel, R. Abstract lb-079: Microbubble mediated focused ultrasound therapy enhances the antitumor potency and durability of anti-PD-L1 checkpoint blockade. Cancer Res. 2020, 80, LB-079. [Google Scholar] [CrossRef]
- Wu, N.; Cao, Y.; Liu, Y.; Zhou, Y.; He, H.; Tang, R.; Wan, L.; Wang, C.; Xiong, X.; Zhong, L.; et al. Low-intensity focused ultrasound targeted microbubble destruction reduces tumor blood supply and sensitizes anti-PD-L1 immunotherapy. Front. Bioeng. Biotechnol. 2023, 11, 1173381. [Google Scholar] [CrossRef]
- Mouratidis, P.X.; Costa, M.; Rivens, I.; Repasky, E.E.; Ter Haar, G. Pulsed focused ultrasound can improve the anti-cancer effects of immune checkpoint inhibitors in murine pancreatic cancer. J. R. Soc. Interface 2021, 18, 20210266. [Google Scholar] [CrossRef]
- Sheybani, N.D.; Breza, V.R.; Paul, S.; McCauley, K.S.; Berr, S.S.; Miller, G.W.; Neumann, K.D.; Price, R.J. ImmunoPET-informed sequence for focused ultrasound-targeted mCD47 blockade controls glioma. J. Control Release 2021, 331, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Tang, J.; Li, H.; Zhou, J.; Tang, N.; Zhu, Q.; Wang, X.; Zhu, B.; Li, N.; Liu, Z. Mechanical destruction using a minimally invasive Ultrasound Needle induces anti-tumor immune responses and synergizes with the anti-PD-L1 blockade. Cancer Lett. 2023, 554, 216009. [Google Scholar] [CrossRef]
- Tan, X.; Yi, C.; Zhang, Y.; Tang, N.; Xu, Y.; Liu, Z. Ultrasound-Targeted Microbubble Destruction Alleviates Immunosuppression Induced by cd71(+) Erythroid Progenitor Cells and Promotes PDL-1 Blockade Immunotherapy in the Lewis Lung Cancer Model. Front. Oncol. 2021, 11, 768222. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Justyniarska, M.; Nowis, D.; Golab, J. Tumor Immune Evasion Induced by Dysregulation of Erythroid Progenitor Cells Development. Cancers 2021, 13, 870. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qiao, Y.D.; Li, X.; Xu, J.L.; Ye, Q.J.; Jiang, N.; Zhang, H.; Wu, X.Y. Intratumoral cd45(+)cd71(+) erythroid cells induce immune tolerance and predict tumor recurrence in hepatocellular carcinoma. Cancer Lett. 2021, 499, 85–98. [Google Scholar] [CrossRef]
- Sundaram, K.M.; Chang, S.S.; Penson, D.F.; Arora, S. Decision Making as a Growth Mechanism in Interventional Oncology: Therapeutic Ultrasound and Prostate Cancer. Semin. Interv. Radiol. 2017, 34, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Khokhlova, T.D.; Cho, C.S.; Khokhlova, V.A. Histotripsy: A Method for Mechanical Tissue Ablation with Ultrasound. Annu. Rev. Biomed. Eng. 2024, 26. [Google Scholar] [CrossRef]
- Rao, R.; Patel, A.; Hanchate, K.; Robinson, E.; Edwards, A.; Shah, S.; Higgins, D.; Haworth, K.J.; Lucke-Wold, B.; Pomeranz Krummel, D.; et al. Advances in Focused Ultrasound for the Treatment of Brain Tumors. Tomography 2023, 9, 1094–1109. [Google Scholar] [CrossRef]
- Siedek, F.; Yeo, S.Y.; Heijman, E.; Grinstein, O.; Bratke, G.; Heneweer, C.; Puesken, M.; Persigehl, T.; Maintz, D.; Grüll, H. Magnetic resonance-guided high-intensity focused ultrasound (mr-hifu): Technical background and overview of current clinical applications (Part 1). In RöFo-Fortschritte auf dem Gebiet der Röntgenstrahlen und der Bildgebenden Verfahren; Georg Thieme Verlag KG: Leipzig, Germany, 2019; pp. 522–530. [Google Scholar]
- Romano, G.; Ventura, E.; Abraham, K.S.; Cersosimo, F.; Giordano, A. Focused ultrasound therapy in cancer care. Ann. Res. Oncol. 2022, 02, 232–245. [Google Scholar] [CrossRef]
- Kooiman, K.; Roovers, S.; Langeveld, S.A.G.; Kleven, R.T.; Dewitte, H.; O’Reilly, M.A.; Escoffre, J.M.; Bouakaz, A.; Verweij, M.D.; Hynynen, K.; et al. Ultrasound-Responsive Cavitation Nuclei for Therapy and Drug Delivery. Ultrasound Med. Biol. 2020, 46, 1296–1325. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; McRae, S.; Cooper, B.; Hu, Y.; Emrick, T.; Pratt, J.; Charles, S.A. End-Functionalized Phosphorylcholine Methacrylates and Their Use in Protein Conjugation. Biomacromolecules 2008, 9, 2891–2897. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Jung, H.; Nam, G.-H.; Kim, I.-S. The right Timing, right combination, right sequence, and right delivery for Cancer immunotherapy. J. Control Release 2021, 331, 321–334. [Google Scholar] [CrossRef]
- DuPage, M.; Jacks, T. Genetically engineered mouse models of cancer reveal new insights about the antitumor immune response. Curr. Opin. Allergy Clin. Immunol. 2013, 25, 192–199. [Google Scholar] [CrossRef]
- DuPage, M.; Cheung, A.F.; Mazumdar, C.; Winslow, M.M.; Bronson, R.; Schmidt, L.M.; Crowley, D.; Chen, J.; Jacks, T. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 2011, 19, 72–85. [Google Scholar] [CrossRef]
- Garbe, A.I.; Vermeer, B.; Gamrekelashvili, J.; Wasielewski, R.V.; Greten, F.R.; Westendorf, A.M.; Buer, J.; Schmid, R.M.; Manns, M.P.; Korangy, F. Genetically induced pancreatic adenocarcinoma is highly immunogenic and causes spontaneous tumor-specific immune responses. Cancer Res. 2006, 66, 508–516. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FUS Modality | Sonication Parameters | Physical Mechanisms and Effects | Biological Effects | Immune Effects | ||
|---|---|---|---|---|---|---|
| Ablative | High Intensity FUS | T-HIFU [62,63,64,65,66] | ISPTA > 1000 W/cm2 P = 3–70 MPa F = 0.2–20 MHz DC% = 100% | -Temperature increase to ~60–85 °C -Stable and inertial cavitation | -Coagulative tissue necrosis -Increase TAAs -Increase DAMPs -Increase lymphatic drainage | -Inflammatory cytokines -Maturity of DCs -Increase TIL population -Heat-shock protein release |
| M-HIFU [66,67,68,69,70] | -Histotripsy ISPTA < 300 W/cm2 P = 20–80 MPa F = 0.2–3 MHz DC% ~0.01% -Boiling histotripsy ISPTA < 600 W/cm2 P = 10–20 MPa F = 0.2–3 MHz DC% ~4% | -Mechanical histotripsy (inertial cavitation, micro-jetting, streaming, and shear stress) -Boiling histotripsy (MB boiling; thermal and mechanical effect, shockwaves) | -Coagulative tissue fractionation and liquefaction -TAA increase -Non-thermally damaged TAA release -Microhemorrhage | -Immunogenic cell death -Better APC activation than HT -TIL increase | ||
| Non-ablative | HT [66,71,72,73,74,75] | ISPTA > 10 W/cm2 P = 0.1–5 MPa F = 0.2–3 MHz DC% = 0.5–100% | -Temperature increase to ~45 °C | -Blood perfusion increase -Tissue oxygenation improvement -Thermally controlled drug release -Protein and DNA damage-induced cell arrest and apoptosis - IFP reduction | -HSP70 increase -Increase in M1 macrophages and NK cells -DC activation -Increase in TNFα and IFNγ -Decrease in MDSCs and IL-10 | |
| Low Intensity FUS | pFUS [76,77,78] | ISPTA < 100 W/cm2 PNP = 0.1–10 MPa F < 3 MHz DC% = 0.5–20% | -Mostly non-thermal effects -Mild heating (<5 degrees) | -Mechanical forces without significant heating -Low cellular damage -Immunomodulation - DNA strand break -Calreticulin translocation | -Expression of CCTFs and CAM -Immune cell homing -Innate and adaptive immune response within the TME (NK cells, DCs, Th1 cells, and CTLs) -Increase in inflammatory immune cells in TDLNs and the spleen | |
| UTMC [67,79] | ISPTA < 10 W/cm2 PNP = 0.1–4 MPa F = 0.2–3 MHz DC% = 0.5–50% | -Sonoporation -Shear stress | -Targeted drug release -Vascular permeabilization -BBB/BTB opening -Reduction in IFP -Reduction in hypoxia -ATP release | -Vascular inflammation -IL-2 and IFNγ increase -TILs -Expression of regulatory genes in MDSCs -Decreased expression of genes responsible for T-cell anergy (i.e., Grail, Itch, Cbib, Grg4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahangiri, S.; Yu, F. Fundamentals and Applications of Focused Ultrasound-Assisted Cancer Immune Checkpoint Inhibition for Solid Tumors. Pharmaceutics 2024, 16, 411. https://doi.org/10.3390/pharmaceutics16030411
Jahangiri S, Yu F. Fundamentals and Applications of Focused Ultrasound-Assisted Cancer Immune Checkpoint Inhibition for Solid Tumors. Pharmaceutics. 2024; 16(3):411. https://doi.org/10.3390/pharmaceutics16030411
Chicago/Turabian StyleJahangiri, Sepideh, and François Yu. 2024. "Fundamentals and Applications of Focused Ultrasound-Assisted Cancer Immune Checkpoint Inhibition for Solid Tumors" Pharmaceutics 16, no. 3: 411. https://doi.org/10.3390/pharmaceutics16030411
APA StyleJahangiri, S., & Yu, F. (2024). Fundamentals and Applications of Focused Ultrasound-Assisted Cancer Immune Checkpoint Inhibition for Solid Tumors. Pharmaceutics, 16(3), 411. https://doi.org/10.3390/pharmaceutics16030411