
Exploring Amino Acid Transporters as Therapeutic Targets for Cancer: An Examination of Inhibitor Structures, Selectivity Issues, and Discovery Approaches



Abstract
1. Introduction
2. Discovering Amino Acid Transporter Inhibitors
3. Alanine, Serine, Cysteine Transporter 2 (ASCT2)
3.1. Background
3.2. ASCT2 Expression and ROLE in Cancer
3.3. Inhibitors of ASCT2

4. Large Neutral Amino Acid Transporter 1 (LAT1)
4.1. Background
4.2. LAT1 Expression and Role in Cancer
4.3. Inhibitors of LAT1

5. Cystine/Glutamate Transporter (xCT)
5.1. Background
5.2. xCT Expression and Role in Cancer
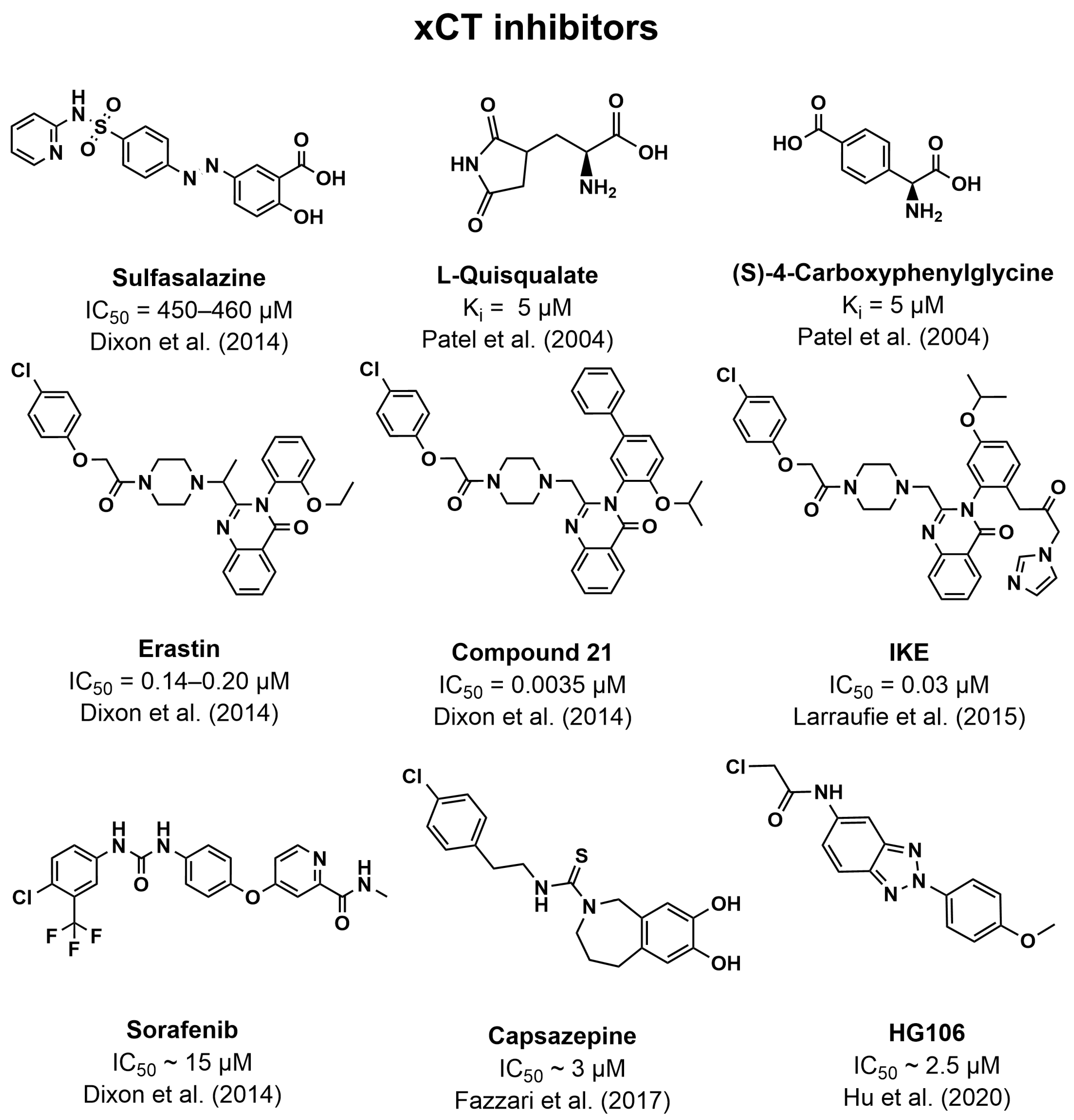
5.3. Inhibitors of xCT

6. Sodium-Coupled Neutral Amino Acid Transporter 1 and 2 (SNAT1/2)
6.1. Background
6.2. SNAT1/SNAT2 Expression and Role in Cancer
6.3. Inhibitors of SNAT1/SNAT2
7. Proton-Coupled Amino Acid Transporter 1 (PAT1)
7.1. Background
7.2. PAT1 Expression and Role in Cancer
7.3. Inhibitors of PAT1
8. Future Perspectives and Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- HUGO Gene Nomenclature Committee (HGNC). HGNC Database. Available online: https://www.genenames.org/tools/search/#!/?query=gene_name:solute&rows=20&start=0&filter=document_type:%22gene%22 (accessed on 28 August 2023).
- Wang, W.W.; Gallo, L.; Jadhav, A.; Hawkins, R.; Parker, C.G. The Druggability of Solute Carriers. J. Med. Chem. 2020, 63, 3834–3867. [Google Scholar] [CrossRef]
- César-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef]
- Sharbeen, G.; McCarroll, J.A.; Akerman, A.; Kopecky, C.; Youkhana, J.; Kokkinos, J.; Holst, J.; Boyer, C.; Erkan, M.; Goldstein, D.; et al. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma Determine Response to SLC7A11 Inhibition. Cancer Res. 2021, 81, 3461–3479. [Google Scholar] [CrossRef]
- Freidman, N.; Chen, I.; Wu, Q.; Briot, C.; Holst, J.; Font, J.; Vandenberg, R.; Ryan, R. Amino Acid Transporters and Exchangers from the SLC1A Family: Structure, Mechanism and Roles in Physiology and Cancer. Neurochem. Res. 2020, 45, 1268–1286. [Google Scholar] [CrossRef]
- Bröer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Bröer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026. [Google Scholar] [CrossRef]
- Sniegowski, T.; Rajasekaran, D.; Sennoune, S.R.; Sunitha, S.; Chen, F.; Fokar, M.; Kshirsagar, S.; Reddy, P.H.; Korac, K.; Mahmud Syed, M.; et al. Amino acid transporter SLC38A5 is a tumor promoter and a novel therapeutic target for pancreatic cancer. Sci. Rep. 2023, 13, 16863. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Yang, S.; Thangaraju, M.; Ganapathy, V. SLC transporters as a novel class of tumour suppressors: Identity, function and molecular mechanisms. Biochem. J. 2016, 473, 1113–1124. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Ooi, A.T.; Gomperts, B.N. Molecular Pathways: Targeting Cellular Energy Metabolism in Cancer via Inhibition of SLC2A1 and LDHA. Clin. Cancer Res. 2015, 21, 2440–2444. [Google Scholar] [CrossRef]
- Higashi, T.; Tamaki, N.; Torizuka, T.; Nakamoto, Y.; Sakahara, H.; Kimura, T.; Honda, T.; Inokuma, T.; Katsushima, S.; Ohshio, G.; et al. FDG uptake, GLUT-1 glucose transporter and cellularity in human pancreatic tumors. J. Nucl. Med. 1998, 39, 1727–1735. [Google Scholar]
- Dai, G.; Levy, O.; Carrasco, N. Cloning and characterization of the thyroid iodide transporter. Nature 1996, 379, 458–460. [Google Scholar] [CrossRef]
- Piert, M.; Shao, X.; Raffel, D.; Davenport, M.S.; Montgomery, J.; Kunju, L.P.; Hockley, B.G.; Siddiqui, J.; Scott, P.J.H.; Chinnaiyan, A.M.; et al. Preclinical Evaluation of (11)C-Sarcosine as a Substrate of Proton-Coupled Amino Acid Transporters and First Human Application in Prostate Cancer. J. Nucl. Med. 2017, 58, 1216–1223. [Google Scholar] [CrossRef]
- Nielsen, C.U.; Krog, N.F.; Sjekirica, I.; Nielsen, S.S.; Pedersen, M.L. SNAT2 is responsible for hyperosmotic induced sarcosine and glycine uptake in human prostate PC-3 cells. Pflugers Arch. 2022, 474, 1249–1262. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef]
- Bröer, A.; Rahimi, F.; Bröer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef]
- Cha, Y.J.; Kim, E.S.; Koo, J.S. Amino Acid Transporters and Glutamine Metabolism in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 907. [Google Scholar] [CrossRef]
- Saito, Y.; Soga, T. Amino acid transporters as emerging therapeutic targets in cancer. Cancer Sci. 2021, 112, 2958–2965. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S. Amino Acid Transporters as Targets for Cancer Therapy: Why, Where, When, and How. Int. J. Mol. Sci. 2020, 21, 6156. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug. Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Zhang, X.; Wright, S.H. Transport Turnover Rates for Human OCT2 and MATE1 Expressed in Chinese Hamster Ovary Cells. Int. J. Mol. Sci. 2022, 23, 1472. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Tatsumi, M.; Groshan, K.; Blakely, R.D.; Richelson, E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 1997, 340, 249–258. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug. Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Dvorak, V.; Wiedmer, T.; Ingles-Prieto, A.; Altermatt, P.; Batoulis, H.; Bärenz, F.; Bender, E.; Digles, D.; Dürrenberger, F.; Heitman, L.H.; et al. An Overview of Cell-Based Assay Platforms for the Solute Carrier Family of Transporters. Front. Pharmacol. 2021, 12, 722889. [Google Scholar] [CrossRef]
- Gauthier-Coles, G.; Bröer, A.; McLeod, M.D.; George, A.J.; Hannan, R.D.; Bröer, S. Identification and characterization of a novel SNAT2 (SLC38A2) inhibitor reveals synergy with glucose transport inhibition in cancer cells. Front. Pharmacol. 2022, 13, 963066. [Google Scholar] [CrossRef]
- Garibsingh, R.A.; Schlessinger, A. Advances and Challenges in Rational Drug Design for SLCs. Trends Pharmacol. Sci. 2019, 40, 790–800. [Google Scholar] [CrossRef]
- Thondorf, I.; Voigt, V.; Schäfer, S.; Gebauer, S.; Zebisch, K.; Laug, L.; Brandsch, M. Three-dimensional quantitative structure-activity relationship analyses of substrates of the human proton-coupled amino acid transporter 1 (hPAT1). Bioorg. Med. Chem. 2011, 19, 6409–6418. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Moesgaard, L.; Pedersen, M.L.; Uhd Nielsen, C.; Kongsted, J. Structure-based discovery of novel P-glycoprotein inhibitors targeting the nucleotide binding domains. Sci. Rep. 2023, 13, 21217. [Google Scholar] [CrossRef]
- Geier, E.G.; Schlessinger, A.; Fan, H.; Gable, J.E.; Irwin, J.J.; Sali, A.; Giacomini, K.M. Structure-based ligand discovery for the Large-neutral Amino Acid Transporter 1, LAT-1. Proc. Natl. Acad. Sci. USA 2013, 110, 5480–5485. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Yee, S.W.; Koleske, M.L.; Zou, L.; Matsson, P.; Chen, E.C.; Kroetz, D.L.; Miller, M.A.; Gozalpour, E.; Chu, X. New and Emerging Research on Solute Carrier and ATP Binding Cassette Transporters in Drug Discovery and Development: Outlook From the International Transporter Consortium. Clin. Pharmacol. Ther. 2022, 112, 540–561. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Christensen, H.N.; Liang, M.; Archer, E.G. A distinct Na+-requiring transport system for alanine, serine, cysteine, and similar amino acids. J. Biol. Chem. 1967, 242, 5237–5246. [Google Scholar] [CrossRef]
- Doyle, F.A.; McGivan, J.D. The bovine renal epithelial cell line NBL-1 expresses a broad specificity Na(+)-dependent neutral amino acid transport system (System Bo) similar to that in bovine renal brush border membrane vesicles. Biochim. Biophys. Acta 1992, 1104, 55–62. [Google Scholar] [CrossRef]
- Bröer, A.; Wagner, C.; Lang, F.; Bröer, S. Neutral amino acid transporter ASCT2 displays substrate-induced Na+ exchange and a substrate-gated anion conductance. Biochem. J. 2000, 346 Pt 3, 705–710. [Google Scholar] [CrossRef]
- Kekuda, R.; Prasad, P.D.; Fei, Y.J.; Torres-Zamorano, V.; Sinha, S.; Yang-Feng, T.L.; Leibach, F.H.; Ganapathy, V. Cloning of the sodium-dependent, broad-scope, neutral amino acid transporter Bo from a human placental choriocarcinoma cell line. J. Biol. Chem. 1996, 271, 18657–18661. [Google Scholar] [CrossRef]
- Arriza, J.L.; Kavanaugh, M.P.; Fairman, W.A.; Wu, Y.N.; Murdoch, G.H.; North, R.A.; Amara, S.G. Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J. Biol. Chem. 1993, 268, 15329–15332. [Google Scholar] [CrossRef]
- Shafqat, S.; Tamarappoo, B.K.; Kilberg, M.S.; Puranam, R.S.; McNamara, J.O.; Guadaño-Ferraz, A.; Fremeau, R.T., Jr. Cloning and expression of a novel Na(+)-dependent neutral amino acid transporter structurally related to mammalian Na+/glutamate cotransporters. J. Biol. Chem. 1993, 268, 15351–15355. [Google Scholar] [CrossRef]
- Torres-Zamorano, V.; Leibach, F.H.; Ganapathy, V. Sodium-dependent homo- and hetero-exchange of neutral amino acids mediated by the amino acid transporter ATB degree. Biochem. Biophys. Res. Commun. 1998, 245, 824–829. [Google Scholar] [CrossRef]
- Bröer, A.; Brookes, N.; Ganapathy, V.; Dimmer, K.S.; Wagner, C.A.; Lang, F.; Bröer, S. The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J. Neurochem. 1999, 73, 2184–2194. [Google Scholar] [PubMed]
- Pingitore, P.; Pochini, L.; Scalise, M.; Galluccio, M.; Hedfalk, K.; Indiveri, C. Large scale production of the active human ASCT2 (SLC1A5) transporter in Pichia pastoris--functional and kinetic asymmetry revealed in proteoliposomes. Biochim. Biophys. Acta 2013, 1828, 2238–2246. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front. Cell. Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Pingitore, P.; Hedfalk, K.; Indiveri, C. Cysteine is not a substrate but a specific modulator of human ASCT2 (SLC1A5) transporter. FEBS Lett. 2015, 589, 3617–3623. [Google Scholar] [CrossRef]
- Mazza, T.; Scalise, M.; Pappacoda, G.; Pochini, L.; Indiveri, C. The involvement of sodium in the function of the human amino acid transporter ASCT2. FEBS Lett. 2021, 595, 3030–3041. [Google Scholar] [CrossRef] [PubMed]
- Garaeva, A.A.; Oostergetel, G.T.; Gati, C.; Guskov, A.; Paulino, C.; Slotboom, D.J. Cryo-EM structure of the human neutral amino acid transporter ASCT2. Nat. Struct. Mol. Biol. 2018, 25, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.W.; Yu, X.J.; Wu, W.C.; Chen, J.; Shi, M.; Zheng, L.; Xu, J. GLUT1 and ASCT2 as Predictors for Prognosis of Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0168907. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Kaira, K.; Tomizawa, Y.; Sunaga, N.; Kawashima, O.; Oriuchi, N.; Tominaga, H.; Nagamori, S.; Kanai, Y.; Yamada, M.; et al. ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. Br. J. Cancer 2014, 110, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.H.; Jung, W.H.; Koo, J.S. Expression of glutamine metabolism-related proteins according to molecular subtype of breast cancer. Endocr. Relat. Cancer 2013, 20, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhao, Y.; Zhao, J.; Wu, S.; Jiang, Y.; Ma, H.; Zhang, T. Upregulated SLC1A5 promotes cell growth and survival in colorectal cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 6006–6014. [Google Scholar] [PubMed]
- Lu, J.; Chen, M.; Tao, Z.; Gao, S.; Li, Y.; Cao, Y.; Lu, C.; Zou, X. Effects of targeting SLC1A5 on inhibiting gastric cancer growth and tumor development in vitro and in vivo. Oncotarget 2017, 8, 76458–76467. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, L.; An, H.; Chang, Y.; Zhang, W.; Zhu, Y.; Xu, L.; Xu, J. High expression of Solute Carrier Family 1, member 5 (SLC1A5) is associated with poor prognosis in clear-cell renal cell carcinoma. Sci. Rep. 2015, 5, 16954. [Google Scholar] [CrossRef]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, R.; Shuai, Y.; Huang, Y.; Jin, R.; Wang, X.; Luo, J. ASCT2 (SLC1A5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br. J. Cancer 2020, 122, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, W.; Ling, Z.; Hu, Q.; Fan, Z.; Cheng, B.; Tao, X. ASCT2 overexpression is associated with poor survival of OSCC patients and ASCT2 knockdown inhibited growth of glutamine-addicted OSCC cells. Cancer Med. 2020, 9, 3489–3499. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, P.J.; Kron, C.D.; Wittke, E.F.; Czerniak, B.N.; Bode, B.P. Targeted Suppression and Knockout of ASCT2 or LAT1 in Epithelial and Mesenchymal Human Liver Cancer Cells Fail to Inhibit Growth. Int. J. Mol. Sci. 2018, 19, 2093. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutté, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Yue, M.; Xiao, D.; Xiu, R.; Gan, L.; Liu, H.; Qing, G. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J. Pathol. 2015, 235, 90–100. [Google Scholar] [CrossRef]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Bungard, C.I.; McGivan, J.D. Glutamine availability up-regulates expression of the amino acid transporter protein ASCT2 in HepG2 cells and stimulates the ASCT2 promoter. Biochem. J. 2004, 382, 27–32. [Google Scholar] [CrossRef]
- Dolińska, M.; Dybel, A.; Hilgier, W.; Zielińska, M.; Zabłocka, B.; Buzańska, L.; Albrecht, J. Glutamine transport in C6 glioma cells: Substrate specificity and modulation in a glutamine deprived culture medium. J. Neurosci. Res. 2001, 66, 959–966. [Google Scholar] [CrossRef]
- Grewer, C.; Grabsch, E. New inhibitors for the neutral amino acid transporter ASCT2 reveal its Na+-dependent anion leak. J. Physiol. 2004, 557, 747–759. [Google Scholar] [CrossRef]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Quek, L.E.; Turner, N.; Freidman, N.; Pang, A.; Guan, Y.F.; Krycer, J.R.; Ryan, R.; Wang, Q.; Holst, J. Benzylserine inhibits breast cancer cell growth by disrupting intracellular amino acid homeostasis and triggering amino acid response pathways. BMC Cancer 2018, 18, 689. [Google Scholar] [CrossRef]
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg. Med. Chem. 2005, 13, 1111–1118. [Google Scholar] [CrossRef]
- Chiu, M.; Sabino, C.; Taurino, G.; Bianchi, M.G.; Andreoli, R.; Giuliani, N.; Bussolati, O. GPNA inhibits the sodium-independent transport system L for neutral amino acids. Amino Acids 2017, 49, 1365–1372. [Google Scholar] [CrossRef]
- Albers, T.; Marsiglia, W.; Thomas, T.; Gameiro, A.; Grewer, C. Defining substrate and blocker activity of alanine-serine-cysteine transporter 2 (ASCT2) Ligands with Novel Serine Analogs. Mol. Pharmacol. 2012, 81, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Oppedisano, F.; Catto, M.; Koutentis, P.A.; Nicolotti, O.; Pochini, L.; Koyioni, M.; Introcaso, A.; Michaelidou, S.S.; Carotti, A.; Indiveri, C. Inactivation of the glutamine/amino acid transporter ASCT2 by 1,2,3-dithiazoles: Proteoliposomes as a tool to gain insights in the molecular mechanism of action and of antitumor activity. Toxicol. Appl. Pharmacol. 2012, 265, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Grewer, C.; Otte, N.J.; Gameiro, A.; Albers, T.; Singh, K.; Shere, H.; Bonomi, M.; Holst, J.; Schlessinger, A. Ligand Discovery for the Alanine-Serine-Cysteine Transporter (ASCT2, SLC1A5) from Homology Modeling and Virtual Screening. PLoS Comput. Biol. 2015, 11, e1004477. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Tanui, R.; Gameiro, A.; Eisenberg, G.; Colas, C.; Schlessinger, A.; Grewer, C. Structure activity relationships of benzylproline-derived inhibitors of the glutamine transporter ASCT2. Bioorg. Med. Chem. Lett. 2017, 27, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.L.; Khodadadi, A.B.; Cuthbertson, M.L.; Smith, J.A.; Manning, H.C. 2-Amino-4-bis(aryloxybenzyl)aminobutanoic acids: A novel scaffold for inhibition of ASCT2-mediated glutamine transport. Bioorg. Med. Chem. Lett. 2016, 26, 1044–1047. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Fairweather, S.; Bröer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front. Pharmacol. 2018, 9, 785. [Google Scholar] [CrossRef]
- Foster, A.C.; Rangel-Diaz, N.; Staubli, U.; Yang, J.Y.; Penjwini, M.; Viswanath, V.; Li, Y.X. Phenylglycine analogs are inhibitors of the neutral amino acid transporters ASCT1 and ASCT2 and enhance NMDA receptor-mediated LTP in rat visual cortex slices. Neuropharmacology 2017, 126, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Reichel, A.; Begley, D.J.; Abbott, N.J. Carrier-mediated delivery of metabotrophic glutamate receptor ligands to the central nervous system: Structural tolerance and potential of the L-system amino acid transporter at the blood-brain barrier. J. Cereb. Blood Flow Metab. 2000, 20, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Garibsingh, R.A.; Otte, N.J.; Ndaru, E.; Colas, C.; Grewer, C.; Holst, J.; Schlessinger, A. Homology Modeling Informs Ligand Discovery for the Glutamine Transporter ASCT2. Front. Chem. 2018, 6, 279. [Google Scholar] [CrossRef] [PubMed]
- Ndaru, E.; Garibsingh, R.A.; Shi, Y.; Wallace, E.; Zakrepine, P.; Wang, J.; Schlessinger, A.; Grewer, C. Novel alanine serine cysteine transporter 2 (ASCT2) inhibitors based on sulfonamide and sulfonic acid ester scaffolds. J. Gen. Physiol. 2019, 151, 357–368. [Google Scholar] [CrossRef]
- Garibsingh, R.A.; Ndaru, E.; Garaeva, A.A.; Shi, Y.; Zielewicz, L.; Zakrepine, P.; Bonomi, M.; Slotboom, D.J.; Paulino, C.; Grewer, C.; et al. Rational design of ASCT2 inhibitors using an integrated experimental-computational approach. Proc. Natl. Acad. Sci. USA 2021, 118, e2104093118. [Google Scholar] [CrossRef] [PubMed]
- Ndaru, E.; Zielewicz, L.; Shi, Y.; Hutchinson, K.; Garibsingh, R.A.; Schlessinger, A.; Grewer, C. Alanine serine cysteine transporter (ASCT) substrate binding site properties probed with hydroxyhomoserine esters. J. Phys. Org. Chem. 2022, 35, e4347. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, J.; Garibsingh, R.A.; Hutchinson, K.; Shi, Y.; Eisenberg, G.; Yu, X.; Schlessinger, A.; Grewer, C. Conserved allosteric inhibition mechanism in SLC1 transporters. eLife 2023, 12, e83464. [Google Scholar] [CrossRef]
- Kanai, Y.; Segawa, H.; Miyamoto, K.; Uchino, H.; Takeda, E.; Endou, H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef]
- Prasad, P.D.; Wang, H.; Huang, W.; Kekuda, R.; Rajan, D.P.; Leibach, F.H.; Ganapathy, V. Human LAT1, a subunit of system L amino acid transporter: Molecular cloning and transport function. Biochem. Biophys. Res. Commun. 1999, 255, 283–288. [Google Scholar] [CrossRef]
- Oxender, D.L.; Christensen, H.N. Evidence for two types of mediation of neutral and amino-acid transport in Ehrlich cells. Nature 1963, 197, 765–767. [Google Scholar] [CrossRef]
- Chien, H.C.; Colas, C.; Finke, K.; Springer, S.; Stoner, L.; Zur, A.A.; Venteicher, B.; Campbell, J.; Hall, C.; Flint, A.; et al. Reevaluating the Substrate Specificity of the L-Type Amino Acid Transporter (LAT1). J. Med. Chem. 2018, 61, 7358–7373. [Google Scholar] [CrossRef]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.; Fukasawa, Y.; et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta 2001, 1514, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.N.; Handlogten, M.E.; Lam, I.; Tager, H.S.; Zand, R. A bicyclic amino acid to improve discriminations among transport systems. J. Biol. Chem. 1969, 244, 1510–1520. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Ristic, Z.; Klauser, S.; Verrey, F. Activation of system L heterodimeric amino acid exchangers by intracellular substrates. Embo. J. 2002, 21, 580–589. [Google Scholar] [CrossRef]
- Boado, R.J.; Li, J.Y.; Nagaya, M.; Zhang, C.; Pardridge, W.M. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 12079–12084. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, T.; Nakamura, M.; Matsuo, A.; Yamasaki, Y.; Takakura, Y.; Hashida, M.; Kanai, Y.; Naito, M.; Tsuruo, T.; Minato, N.; et al. The 4F2hc/LAT1 complex transports L-DOPA across the blood-brain barrier. Brain Res. 2000, 879, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Ecker, G.F. Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1. Int. J. Mol. Sci. 2018, 19, 1278. [Google Scholar] [CrossRef] [PubMed]
- Sang, J.; Lim, Y.P.; Panzica, M.; Finch, P.; Thompson, N.L. TA1, a highly conserved oncofetal complementary DNA from rat hepatoma, encodes an integral membrane protein associated with liver development, carcinogenesis, and cell activation. Cancer Res. 1995, 55, 1152–1159. [Google Scholar] [PubMed]
- Betsunoh, H.; Fukuda, T.; Anzai, N.; Nishihara, D.; Mizuno, T.; Yuki, H.; Masuda, A.; Yamaguchi, Y.; Abe, H.; Yashi, M.; et al. Increased expression of system large amino acid transporter (LAT)-1 mRNA is associated with invasive potential and unfavorable prognosis of human clear cell renal cell carcinoma. BMC Cancer 2013, 13, 509. [Google Scholar] [CrossRef]
- Hayase, S.; Kumamoto, K.; Saito, K.; Kofunato, Y.; Sato, Y.; Okayama, H.; Miyamoto, K.; Ohki, S.; Takenoshita, S. L-type amino acid transporter 1 expression is upregulated and associated with cellular proliferation in colorectal cancer. Oncol. Lett. 2017, 14, 7410–7416. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Miyamoto, M.; Takano, M.; Tsuda, H. Correlation of high LAT1 expression with the prognosis of endometrioid carcinoma of the uterine corpus. Virchows Arch. 2020, 477, 421–427. [Google Scholar] [CrossRef]
- Wang, J.; Fei, X.; Wu, W.; Chen, X.; Su, L.; Zhu, Z.; Zhou, Y. SLC7A5 Functions as a Downstream Target Modulated by CRKL in Metastasis Process of Gastric Cancer SGC-7901 Cells. PLoS ONE 2016, 11, e01661472016. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Sakamoto, S.; Matsushima, J.; Kimura, T.; Ueda, T.; Mizokami, A.; Kanai, Y.; Ichikawa, T. Up-Regulation of LAT1 during Antiandrogen Therapy Contributes to Progression in Prostate Cancer Cells. J. Urol. 2016, 195, 1588–1597. [Google Scholar] [CrossRef]
- Kobayashi, H.; Ishii, Y.; Takayama, T. Expression of L-type amino acid transporter 1 (LAT1) in esophageal carcinoma. J. Surg. Oncol. 2005, 90, 233–238. [Google Scholar] [CrossRef]
- Takeuchi, K.; Ogata, S.; Nakanishi, K.; Ozeki, Y.; Hiroi, S.; Tominaga, S.; Aida, S.; Matsuo, H.; Sakata, T.; Kawai, T. LAT1 expression in non-small-cell lung carcinomas: Analyses by semiquantitative reverse transcription-PCR (237 cases) and immunohistochemistry (295 cases). Lung Cancer 2010, 68, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Sunose, Y.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; Itoh, H.; Nagamori, S.; et al. Prognostic significance of L-type amino-acid transporter 1 expression in surgically resected pancreatic cancer. Br. J. Cancer 2012, 107, 632–638. [Google Scholar] [CrossRef]
- Honjo, H.; Kaira, K.; Miyazaki, T.; Yokobori, T.; Kanai, Y.; Nagamori, S.; Oyama, T.; Asao, T.; Kuwano, H. Clinicopathological significance of LAT1 and ASCT2 in patients with surgically resected esophageal squamous cell carcinoma. J. Surg. Oncol. 2016, 113, 381–389. [Google Scholar] [CrossRef]
- Ichinoe, M.; Mikami, T.; Yoshida, T.; Igawa, I.; Tsuruta, T.; Nakada, N.; Anzai, N.; Suzuki, Y.; Endou, H.; Okayasu, I. High expression of L-type amino-acid transporter 1 (LAT1) in gastric carcinomas: Comparison with non-cancerous lesions. Pathol. Int. 2011, 61, 281–289. [Google Scholar] [CrossRef]
- Isoda, A.; Kaira, K.; Iwashina, M.; Oriuchi, N.; Tominaga, H.; Nagamori, S.; Kanai, Y.; Oyama, T.; Asao, T.; Matsumoto, M.; et al. Expression of L-type amino acid transporter 1 (LAT1) as a prognostic and therapeutic indicator in multiple myeloma. Cancer Sci. 2014, 105, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Kanai, Y.; Choi, H.W.; Tangtrongsup, S.; Chairoungdua, A.; Babu, E.; Tachampa, K.; Anzai, N.; Iribe, Y.; Endou, H. Characterization of the system L amino acid transporter in T24 human bladder carcinoma cells. Biochim. Biophys. Acta 2002, 1565, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, M.; Ohno, Y.; Masuko, K.; Takeuchi, A.; Suda, K.; Kubo, A.; Kawahara, R.; Okazaki, S.; Tanaka, T.; Saya, H.; et al. Oncogenicity of L-type amino-acid transporter 1 (LAT1) revealed by targeted gene disruption in chicken DT40 cells: LAT1 is a promising molecular target for human cancer therapy. Biochem. Biophys. Res. Commun. 2011, 406, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Haining, Z.; Kawai, N.; Miyake, K.; Okada, M.; Okubo, S.; Zhang, X.; Fei, Z.; Tamiya, T. Relation of LAT1/4F2hc expression with pathological grade, proliferation and angiogenesis in human gliomas. BMC Clin. Pathol. 2012, 12, 4. [Google Scholar] [CrossRef]
- Quan, L.; Ohgaki, R.; Hara, S.; Okuda, S.; Wei, L.; Okanishi, H.; Nagamori, S.; Endou, H.; Kanai, Y. Amino acid transporter LAT1 in tumor-associated vascular endothelium promotes angiogenesis by regulating cell proliferation and VEGF-A-dependent mTORC1 activation. J. Exp. Clin. Cancer Res. 2020, 39, 266. [Google Scholar] [CrossRef]
- Wiriyasermkul, P.; Nagamori, S.; Tominaga, H.; Oriuchi, N.; Kaira, K.; Nakao, H.; Kitashoji, T.; Ohgaki, R.; Tanaka, H.; Endou, H.; et al. Transport of 3-fluoro-L-α-methyl-tyrosine by tumor-upregulated L-type amino acid transporter 1: A cause of the tumor uptake in PET. J. Nucl. Med. 2012, 53, 1253–1261. [Google Scholar] [CrossRef]
- Nobusawa, A.; Kim, M.; Kaira, K.; Miyashita, G.; Negishi, A.; Oriuchi, N.; Higuchi, T.; Tsushima, Y.; Kanai, Y.; Yokoo, S.; et al. Diagnostic usefulness of ¹⁸F-FAMT PET and L-type amino acid transporter 1 (LAT1) expression in oral squamous cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 1692–1700. [Google Scholar] [CrossRef]
- Christensen, H.N. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol. Rev. 1990, 70, 43–77. [Google Scholar] [CrossRef]
- Ohshima, Y.; Kaira, K.; Yamaguchi, A.; Oriuchi, N.; Tominaga, H.; Nagamori, S.; Kanai, Y.; Yokobori, T.; Miyazaki, T.; Asao, T.; et al. Efficacy of system l amino acid transporter 1 inhibition as a therapeutic target in esophageal squamous cell carcinoma. Cancer Sci. 2016, 107, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Sunose, Y.; Ohshima, Y.; Ishioka, N.S.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; et al. Clinical significance of L-type amino acid transporter 1 expression as a prognostic marker and potential of new targeting therapy in biliary tract cancer. BMC Cancer 2013, 13, 482. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Kaira, K.; Oriuchi, N.; Shimizu, K.; Tominaga, H.; Yanagitani, N.; Sunaga, N.; Ishizuka, T.; Nagamori, S.; Promchan, K.; et al. Inhibition of L-type amino acid transporter 1 has antitumor activity in non-small cell lung cancer. Anticancer Res. 2010, 30, 4819–4828. [Google Scholar] [PubMed]
- Segawa, H.; Fukasawa, Y.; Miyamoto, K.; Takeda, E.; Endou, H.; Kanai, Y. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J. Biol. Chem. 1999, 274, 19745–19751. [Google Scholar] [CrossRef]
- Babu, E.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Iribe, Y.; Tangtrongsup, S.; Jutabha, P.; Li, Y.; Ahmed, N.; Sakamoto, S.; et al. Identification of a novel system L amino acid transporter structurally distinct from heterodimeric amino acid transporters. J. Biol. Chem. 2003, 278, 43838–43845. [Google Scholar] [CrossRef] [PubMed]
- Bodoy, S.; Martín, L.; Zorzano, A.; Palacín, M.; Estévez, R.; Bertran, J. Identification of LAT4, a novel amino acid transporter with system L activity. J. Biol. Chem. 2005, 280, 12002–12011. [Google Scholar] [CrossRef] [PubMed]
- Sloan, J.L.; Mager, S. Cloning and functional expression of a human Na(+) and Cl(-)-dependent neutral and cationic amino acid transporter B(0+). J. Biol. Chem. 1999, 274, 23740–23745. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Klingel, K.; Kowalczuk, S.; Rasko, J.E.; Cavanaugh, J.; Bröer, S. Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J. Biol. Chem. 2004, 279, 24467–24476. [Google Scholar] [CrossRef]
- Uchino, H.; Kanai, Y.; Kim, D.K.; Wempe, M.F.; Chairoungdua, A.; Morimoto, E.; Anders, M.W.; Endou, H. Transport of amino acid-related compounds mediated by L-type amino acid transporter 1 (LAT1): Insights into the mechanisms of substrate recognition. Mol. Pharmacol. 2002, 61, 729–737. [Google Scholar] [CrossRef]
- Nagamori, S.; Wiriyasermkul, P.; Okuda, S.; Kojima, N.; Hari, Y.; Kiyonaka, S.; Mori, Y.; Tominaga, H.; Ohgaki, R.; Kanai, Y. Structure-activity relations of leucine derivatives reveal critical moieties for cellular uptake and activation of mTORC1-mediated signaling. Amino Acids 2016, 48, 1045–1058. [Google Scholar] [CrossRef]
- Su, T.Z.; Feng, M.R.; Weber, M.L. Mediation of highly concentrative uptake of pregabalin by L-type amino acid transport in Chinese hamster ovary and Caco-2 cells. J. Pharmacol. Exp. Ther. 2005, 313, 1406–1415. [Google Scholar] [CrossRef]
- Augustyn, E.; Finke, K.; Zur, A.A.; Hansen, L.; Heeren, N.; Chien, H.C.; Lin, L.; Giacomini, K.M.; Colas, C.; Schlessinger, A.; et al. LAT-1 activity of meta-substituted phenylalanine and tyrosine analogs. Bioorg. Med. Chem. Lett. 2016, 26, 2616–2621. [Google Scholar] [CrossRef]
- Kongpracha, P.; Nagamori, S.; Wiriyasermkul, P.; Tanaka, Y.; Kaneda, K.; Okuda, S.; Ohgaki, R.; Kanai, Y. Structure-activity relationship of a novel series of inhibitors for cancer type transporter L-type amino acid transporter 1 (LAT1). J. Pharmacol. Sci. 2017, 133, 96–102. [Google Scholar] [CrossRef]
- Zur, A.A.; Chien, H.C.; Augustyn, E.; Flint, A.; Heeren, N.; Finke, K.; Hernandez, C.; Hansen, L.; Miller, S.; Lin, L.; et al. LAT1 activity of carboxylic acid bioisosteres: Evaluation of hydroxamic acids as substrates. Bioorg. Med. Chem. Lett. 2016, 26, 5000–5006. [Google Scholar] [CrossRef] [PubMed]
- Ylikangas, H.; Malmioja, K.; Peura, L.; Gynther, M.; Nwachukwu, E.O.; Leppänen, J.; Laine, K.; Rautio, J.; Lahtela-Kakkonen, M.; Huttunen, K.M.; et al. Quantitative insight into the design of compounds recognized by the L-type amino acid transporter 1 (LAT1). ChemMedChem 2014, 9, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Scalise, M.; Galluccio, M.; Wieder, M.; Seidel, T.; Langer, T.; Indiveri, C.; Ecker, G.F. Discovery of Potent Inhibitors for the Large Neutral Amino Acid Transporter 1 (LAT1) by Structure-Based Methods. Int. J. Mol. Sci. 2018, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Müller, J.; Sadurní, A.; Rubin, M.; Canivete Cuissa, I.A.; Keller, C.; Hartmann, M.; Singer, S.; Gertsch, J.; Altmann, K.H. The Evaluation of l-Tryptophan Derivatives as Inhibitors of the l-Type Amino Acid Transporter LAT1 (SLC7A5). ChemMedChem 2022, 17, e202200308. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Hosoda, N.; Endo, H.; Saito, K.; Tsujihara, K.; Yamamura, M.; Sakata, T.; Anzai, N.; Wempe, M.F.; Kanai, Y.; et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 2010, 101, 173–179. [Google Scholar] [CrossRef]
- Yun, D.W.; Lee, S.A.; Park, M.G.; Kim, J.S.; Yu, S.K.; Park, M.R.; Kim, S.G.; Oh, J.S.; Kim, C.S.; Kim, H.J.; et al. JPH203, an L-type amino acid transporter 1-selective compound, induces apoptosis of YD-38 human oral cancer cells. J. Pharmacol. Sci. 2014, 124, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Jutabha, P.; Maeda, S.; Supak, Y.; Ouchi, M.; Endou, H.; Fujita, T.; Chida, M.; Anzai, N. LAT1 acts as a crucial transporter of amino acids in human thymic carcinoma cells. J. Pharmacol. Sci. 2016, 132, 201–204. [Google Scholar] [CrossRef]
- Yothaisong, S.; Dokduang, H.; Anzai, N.; Hayashi, K.; Namwat, N.; Yongvanit, P.; Sangkhamanon, S.; Jutabha, P.; Endou, H.; Loilome, W. Inhibition of l-type amino acid transporter 1 activity as a new therapeutic target for cholangiocarcinoma treatment. Tumour Biol. 2017, 39, 1010428317694545. [Google Scholar] [CrossRef]
- Choi, D.W.; Kim, D.K.; Kanai, Y.; Wempe, M.F.; Endou, H.; Kim, J.K. JPH203, a selective L-type amino acid transporter 1 inhibitor, induces mitochondria-dependent apoptosis in Saos2 human osteosarcoma cells. Korean J. Physiol. Pharmacol. 2017, 21, 599–607. [Google Scholar] [CrossRef]
- Häfliger, P.; Graff, J.; Rubin, M.; Stooss, A.; Dettmer, M.S.; Altmann, K.H.; Gertsch, J.; Charles, R.P. The LAT1 inhibitor JPH203 reduces growth of thyroid carcinoma in a fully immunocompetent mouse model. J. Exp. Clin. Cancer Res. 2018, 37, 234. [Google Scholar] [CrossRef]
- Higuchi, K.; Sakamoto, S.; Ando, K.; Maimaiti, M.; Takeshita, N.; Okunushi, K.; Reien, Y.; Imamura, Y.; Sazuka, T.; Nakamura, K.; et al. Characterization of the expression of LAT1 as a prognostic indicator and a therapeutic target in renal cell carcinoma. Sci. Rep. 2019, 9, 16776. [Google Scholar] [CrossRef]
- Cormerais, Y.; Pagnuzzi-Boncompagni, M.; Schrötter, S.; Giuliano, S.; Tambutté, E.; Endou, H.; Wempe, M.F.; Pagès, G.; Pouysségur, J.; Picco, V. Inhibition of the amino-acid transporter LAT1 demonstrates anti-neoplastic activity in medulloblastoma. J. Cell. Mol. Med. 2019, 23, 2711–2718. [Google Scholar] [CrossRef] [PubMed]
- Muto, Y.; Furihata, T.; Kaneko, M.; Higuchi, K.; Okunushi, K.; Morio, H.; Reien, Y.; Uesato, M.; Matsubara, H.; Anzai, N. Different Response Profiles of Gastrointestinal Cancer Cells to an L-Type Amino Acid Transporter Inhibitor, JPH203. Anticancer Res. 2019, 39, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Maimaiti, M.; Sakamoto, S.; Yamada, Y.; Sugiura, M.; Rii, J.; Takeuchi, N.; Imamura, Y.; Furihata, T.; Ando, K.; Higuchi, K.; et al. Expression of L-type amino acid transporter 1 as a molecular target for prognostic and therapeutic indicators in bladder carcinoma. Sci. Rep. 2020, 10, 1292. [Google Scholar] [CrossRef] [PubMed]
- Satou, M.; Wang, J.; Nakano-Tateno, T.; Teramachi, M.; Suzuki, T.; Hayashi, K.; Lamothe, S.; Hao, Y.; Kurata, H.; Sugimoto, H.; et al. L-type amino acid transporter 1, LAT1, in growth hormone-producing pituitary tumor cells. Mol. Cell. Endocrinol. 2020, 515, 110868. [Google Scholar] [CrossRef] [PubMed]
- Jigjidkhorloo, N.; Kanekura, K.; Matsubayashi, J.; Akahane, D.; Fujita, K.; Oikawa, K.; Kurata, A.; Takanashi, M.; Endou, H.; Nagao, T.; et al. Expression of L-type amino acid transporter 1 is a poor prognostic factor for Non-Hodgkin’s lymphoma. Sci. Rep. 2021, 11, 21638. [Google Scholar] [CrossRef] [PubMed]
- Nishikubo, K.; Ohgaki, R.; Okanishi, H.; Okuda, S.; Xu, M.; Endou, H.; Kanai, Y. Pharmacologic inhibition of LAT1 predominantly suppresses transport of large neutral amino acids and downregulates global translation in cancer cells. J. Cell. Mol. Med. 2022, 26, 5246–5256. [Google Scholar] [CrossRef] [PubMed]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Invest. New. Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef]
- Bo, T.; Kobayashi, S.; Inanami, O.; Fujii, J.; Nakajima, O.; Ito, T.; Yasui, H. LAT1 inhibitor JPH203 sensitizes cancer cells to radiation by enhancing radiation-induced cellular senescence. Transl. Oncol. 2021, 14, 101212. [Google Scholar] [CrossRef]
- Napolitano, L.; Scalise, M.; Koyioni, M.; Koutentis, P.; Catto, M.; Eberini, I.; Parravicini, C.; Palazzolo, L.; Pisani, L.; Galluccio, M.; et al. Potent inhibitors of human LAT1 (SLC7A5) transporter based on dithiazole and dithiazine compounds for development of anticancer drugs. Biochem. Pharmacol. 2017, 143, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Prejanò, M.; Romeo, I.; La Serra, M.A.; Russo, N.; Marino, T. Computational Study Reveals the Role of Water Molecules in the Inhibition Mechanism of LAT1 by 1,2,3-Dithiazoles. J. Chem. Inf. Model. 2021, 61, 5883–5892. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S.; Kitamura, E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J. Biol. Chem. 1980, 255, 2372–2376. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar] [CrossRef] [PubMed]
- Makowske, M.; Christensen, H.N. Contrasts in transport systems for anionic amino acids in hepatocytes and a hepatoma cell line HTC. J. Biol. Chem. 1982, 257, 5663–5670. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Kuriyama-Matsumura, K.; Okuno, S.; Bannai, S. Molecular cloning and expression of human xCT, the light chain of amino acid transport system xc. Antioxid. Redox Signal 2000, 2, 665–671. [Google Scholar] [CrossRef]
- Parker, J.L.; Deme, J.C.; Kolokouris, D.; Kuteyi, G.; Biggin, P.C.; Lea, S.M.; Newstead, S. Molecular basis for redox control by the human cystine/glutamate antiporter system xc−. Nat. Commun. 2021, 12, 7147. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Tissue Expression of SLC7A11. Available online: https://www.proteinatlas.org/ENSG00000151012-SLC7A11/tissue (accessed on 28 August 2023).
- Pampliega, O.; Domercq, M.; Soria, F.N.; Villoslada, P.; Rodríguez-Antigüedad, A.; Matute, C. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J. Neuroinflamm. 2011, 8, 63. [Google Scholar] [CrossRef]
- Eagle, H. The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J. Exp. Med. 1955, 102, 37–48. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Bannai, S.; Tsukeda, H.; Okumura, H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem. Biophys. Res. Commun. 1977, 74, 1582–1588. [Google Scholar] [CrossRef]
- Meister, A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol. Ther. 1991, 51, 155–194. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Bannai, S.; Kitamura, E. Adaptive enhancement of cystine and glutamate uptake in human diploid fibroblasts in culture. Biochim. Biophys. Acta 1982, 721, 1–10. [Google Scholar] [CrossRef]
- Sato, H.; Nomura, S.; Maebara, K.; Sato, K.; Tamba, M.; Bannai, S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem. Biophys. Res. Commun. 2004, 325, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, Y.; Wu, H. A novel scoring system for acute myeloid leukemia risk assessment based on the expression levels of six genes. Int. J. Mol. Med. 2018, 42, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van ‘t Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Yi, S.; Wei, L.; Zhao, B.; Li, J.; Cai, X.; Dong, C.; Liu, X. Microarray-based identification of genes associated with prognosis and drug resistance in ovarian cancer. J. Cell. Biochem. 2019, 120, 6057–6070. [Google Scholar] [CrossRef]
- Sugano, K.; Maeda, K.; Ohtani, H.; Nagahara, H.; Shibutani, M.; Hirakawa, K. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 2015, 35, 677–682. [Google Scholar]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef]
- Zhong, W.; Weiss, H.L.; Jayswal, R.D.; Hensley, P.J.; Downes, L.M.; St Clair, D.K.; Chaiswing, L. Extracellular redox state shift: A novel approach to target prostate cancer invasion. Free Radic. Biol. Med. 2018, 117, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Okabe, H.; Beppu, T.; Chikamoto, A.; Hayashi, H.; Imai, K.; Mima, K.; Nakagawa, S.; Ishimoto, T.; Miyake, K.; et al. Cystine/glutamic acid transporter is a novel marker for predicting poor survival in patients with hepatocellular carcinoma. Oncol. Rep. 2013, 29, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.M.; Buckingham, S.C.; Campbell, S.L.; Robel, S.; Holt, K.T.; Ogunrinu-Babarinde, T.; Warren, P.P.; White, D.M.; Reid, M.A.; Eschbacher, J.M.; et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 2015, 7, 289ra286. [Google Scholar] [CrossRef]
- Shin, S.S.; Jeong, B.S.; Wall, B.A.; Li, J.; Shan, N.L.; Wen, Y.; Goydos, J.S.; Chen, S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 2018, 7, 86. [Google Scholar] [CrossRef]
- Lim, J.; Delaidelli, A.; Minaker, S.; Zhang, H.-F.; Colovic, M.; Yang, H.; Negri, G.; Von Karstedt, S.; Lockwood, W.; Schaffer, P.; et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 116, 201821323. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Okuno, S.; Sato, H.; Kuriyama-Matsumura, K.; Tamba, M.; Wang, H.; Sohda, S.; Hamada, H.; Yoshikawa, H.; Kondo, T.; Bannai, S. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br. J. Cancer 2003, 88, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Ling, V.; Wang, Y.Z.; Gout, P.W. The xc- cystine/glutamate antiporter: A mediator of pancreatic cancer growth with a role in drug resistance. Br. J. Cancer 2008, 99, 464–472. [Google Scholar] [CrossRef]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadée, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef] [PubMed]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased Expression of System xc- in Glioblastoma Confers an Altered Metabolic State and Temozolomide Resistance. Mol. Cancer Res. 2016, 14, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 2017, 6, e27713. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell. Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)− cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef]
- Doxsee, D.W.; Gout, P.W.; Kurita, T.; Lo, M.; Buckley, A.R.; Wang, Y.; Xue, H.; Karp, C.M.; Cutz, J.C.; Cunha, G.R.; et al. Sulfasalazine-induced cystine starvation: Potential use for prostate cancer therapy. Prostate 2007, 67, 162–171. [Google Scholar] [CrossRef]
- Ogihara, K.; Kikuchi, E.; Okazaki, S.; Hagiwara, M.; Takeda, T.; Matsumoto, K.; Kosaka, T.; Mikami, S.; Saya, H.; Oya, M. Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer. Cancer Sci. 2019, 110, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Patel, D.; Kharkar, P.S.; Gandhi, N.S.; Kaur, E.; Dutt, S.; Nandave, M. Novel analogs of sulfasalazine as system x(c)(-) antiporter inhibitors: Insights from the molecular modeling studies. Drug Dev. Res. 2019, 80, 758–777. [Google Scholar] [CrossRef]
- Cirillo, D.; Sarowar, S.; Øyvind Enger, P.; Bjørsvik, H.R. Structure-Activity-Relationship-Aided Design and Synthesis of xCT Antiporter Inhibitors. ChemMedChem 2021, 16, 2650–2668. [Google Scholar] [CrossRef]
- Patel, S.A.; Warren, B.A.; Rhoderick, J.F.; Bridges, R.J. Differentiation of substrate and non-substrate inhibitors of transport system xc(-): An obligate exchanger of L-glutamate and L-cystine. Neuropharmacology 2004, 46, 273–284. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Larraufie, M.H.; Yang, W.S.; Jiang, E.; Thomas, A.G.; Slusher, B.S.; Stockwell, B.R. Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg. Med. Chem. Lett. 2015, 25, 4787–4792. [Google Scholar] [CrossRef]
- Zheng, J.; Sato, M.; Mishima, E.; Sato, H.; Proneth, B.; Conrad, M. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, J.; Balenko, M.D.; Zacal, N.; Singh, G. Identification of capsazepine as a novel inhibitor of system x(c)(-) and cancer-induced bone pain. J. Pain. Res. 2017, 10, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.N.; Oxender, D.L.; Liang, M.; Vatz, K.A. The Use of N-Methylation to Direct the Route of Mediated Transport of Amino Acids. J. Biol. Chem. 1965, 240, 3609–3616. [Google Scholar] [CrossRef] [PubMed]
- Varoqui, H.; Yao, D.; Zhu, H.; Ming, H.; Erickson, J.D. Cloning and Functional Identification of a Neuronal Glutamine Transporter. J. Biol. Chem. 2000, 275, 4049–4054. [Google Scholar] [CrossRef]
- Reimer, R.J.; Chaudhry, F.A.; Gray, A.T.; Edwards, R.H. Amino acid transport system A resembles system N in sequence but differs in mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 7715–7720. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Nakanishi, T.; Fei, Y.J.; Huang, W.; Ganapathy, M.E.; Leibach, F.H.; Ganapathy, V. Cloning of an amino acid transporter with functional characteristics and tissue expression pattern identical to that of system A. J. Biol. Chem. 2000, 275, 16473–16477. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Mackenzie, B.; Ming, H.; Varoqui, H.; Zhu, H.; Hediger, M.A.; Erickson, J.D. A novel system A isoform mediating Na+/neutral amino acid cotransport. J. Biol. Chem. 2000, 275, 22790–22797. [Google Scholar] [CrossRef] [PubMed]
- Albers, A.; Bröer, A.; Wagner, C.A.; Setiawan, I.; Lang, P.A.; Kranz, E.U.; Lang, F.; Bröer, S. Na+ transport by the neural glutamine transporter ATA1. Pflügers Archiv. 2001, 443, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, B.; Schäfer, M.K.H.; Erickson, J.D.; Hediger, M.A.; Weihe, E.; Varoqui, H. Functional Properties and Cellular Distribution of the System A Glutamine Transporter SNAT1 Support Specialized Roles in Central Neurons. J. Biol. Chem. 2003, 278, 23720–23730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Grewer, C. The Sodium-Coupled Neutral Amino Acid Transporter SNAT2 Mediates an Anion Leak Conductance that Is Differentially Inhibited by Transported Substrates. Biophys. J. 2007, 92, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, B.; Erickson, J.D. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch. 2004, 447, 784–795. [Google Scholar] [CrossRef]
- Zhang, Z.; Albers, T.; Fiumera, H.L.; Gameiro, A.; Grewer, C. A Conserved Na+ Binding Site of the Sodium-coupled Neutral Amino Acid Transporter 2 (SNAT2). J. Biol. Chem. 2009, 284, 25314–25323. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.; Sugawara, M.; Devoe, L.D.; Leibach, F.H.; Prasad, P.D.; Ganapathy, V. Cloning and functional expression of ATA1, a subtype of amino acid transporter A, from human placenta. Biochem. Biophys. Res. Commun. 2000, 273, 1175–1179. [Google Scholar] [CrossRef]
- Hatanaka, T.; Huang, W.; Wang, H.; Sugawara, M.; Prasad, P.D.; Leibach, F.H.; Ganapathy, V. Primary structure, functional characteristics and tissue expression pattern of human ATA2, a subtype of amino acid transport system A. Biochim. Biophys. Acta 2000, 1467, 1–6. [Google Scholar] [CrossRef]
- Gazzola, G.C.; Franchi, R.; Saibene, V.; Ronchi, P.; Guidotti, G.G. Regulation of amino acid transport in chick embryo heart cells. I. Adaptive system of mediation for neutral amino acids. Biochim. Biophys. Acta 1972, 266, 407–421. [Google Scholar] [CrossRef]
- Riggs, T.R.; Pan, M.W. Transport of amino acids into the oestrogen-primed uterus. Enchancement of the uptake by a preliminary incubation. Biochem. J. 1972, 128, 19–27. [Google Scholar] [CrossRef]
- Gazzola, R.F.; Sala, R.; Bussolati, O.; Visigalli, R.; Dall’Asta, V.; Ganapathy, V.; Gazzola, G.C. The adaptive regulation of amino acid transport system A is associated to changes in ATA2 expression. FEBS Lett. 2001, 490, 11–14. [Google Scholar] [CrossRef]
- Palii, S.S.; Chen, H.; Kilberg, M.S. Transcriptional control of the human sodium-coupled neutral amino acid transporter system A gene by amino acid availability is mediated by an intronic element. J. Biol. Chem. 2004, 279, 3463–3471. [Google Scholar] [CrossRef] [PubMed]
- Gaccioli, F.; Huang, C.C.; Wang, C.; Bevilacqua, E.; Franchi-Gazzola, R.; Gazzola, G.C.; Bussolati, O.; Snider, M.D.; Hatzoglou, M. Amino acid starvation induces the SNAT2 neutral amino acid transporter by a mechanism that involves eukaryotic initiation factor 2alpha phosphorylation and cap-independent translation. J. Biol. Chem. 2006, 281, 17929–17940. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, H.; Yamazaki, K.; Takekuma, Y.; Ganapathy, V.; Sugawara, M. Regulatory mechanisms of SNAT2, an amino acid transporter, in L6 rat skeletal muscle cells by insulin, osmotic shock and amino acid deprivation. Amino Acids 2009, 36, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Petronini, P.G.; De Angelis, E.; Borghetti, A.F.; Wheeler, K.P. Osmotically inducible uptake of betaine via amino acid transport system A in SV-3T3 cells. Biochem. J. 1994, 300, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Tramacere, M.; Petronini, P.G.; Borghetti, A.F. Effect of hyperosmolarity on the activity of amino acid transport system L in avian fibroblasts. J. Cell. Physiol. 1984, 121, 81–86. [Google Scholar] [CrossRef]
- Franchi-Gazzola, R.; Dall’Asta, V.; Sala, R.; Visigalli, R.; Bevilacqua, E.; Gaccioli, F.; Gazzola, G.C.; Bussolati, O. The role of the neutral amino acid transporter SNAT2 in cell volume regulation. Acta Physiol. 2006, 187, 273–283. [Google Scholar] [CrossRef]
- Gazzola, G.C.; Dall’Asta, V.; Nucci, F.A.; Rossi, P.A.; Bussolati, O.; Hoffmann, E.K.; Guidotti, G.G. Role of Amino Acid Transport System A in the Control of Cell Volume in Cultured Human Fibroblasts. Cell. Physiol. Biochem. 1991, 1, 131–142. [Google Scholar] [CrossRef]
- Dall’Asta, V.; Bussolati, O.; Sala, R.; Parolari, A.; Alamanni, F.; Biglioli, P.; Gazzola, G.C. Amino acids are compatible osmolytes for volume recovery after hypertonic shrinkage in vascular endothelial cells. Am. J. Physiol. 1999, 276, C865–C872. [Google Scholar] [CrossRef]
- Dall’Asta, V.; Rossi, P.A.; Bussolati, O.; Gazzola, G.C. Regulatory volume decrease of cultured human fibroblasts involves changes in intracellular amino-acid pool. Biochim. Biophys. Acta 1994, 1220, 139–145. [Google Scholar] [CrossRef]
- Fan, S.J.; Goberdhan, D.C.I. PATs and SNATs: Amino Acid Sensors in Disguise. Front. Pharmacol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Hundal, H.S. SNAT2 transceptor signalling via mTOR A role in cell growth and proliferation. Front. Biosci. 2011, E3, 1289–1299. [Google Scholar] [CrossRef]
- Lei, H.T.; Mu, X.; Hattne, J.; Gonen, T. A conformational change in the N terminus of SLC38A9 signals mTORC1 activation. Structure 2021, 29, 426–432.e8. [Google Scholar] [CrossRef]
- Kondoh, N.; Imazeki, N.; Arai, M.; Hada, A.; Hatsuse, K.; Matsuo, H.; Matsubara, O.; Ohkura, S.; Yamamoto, M. Activation of a system A amino acid transporter, ATA1/SLC38A1, in human hepatocellular carcinoma and preneoplastic liver tissues. Int. J. Oncol. 2007, 31, 81–87. [Google Scholar] [CrossRef][Green Version]
- Böhme-Schäfer, I.; Lörentz, S.; Bosserhoff, A.K. Role of Amino Acid Transporter SNAT1/SLC38A1 in Human Melanoma. Cancers 2022, 14, 2151. [Google Scholar] [CrossRef]
- Morotti, M.; Zois, C.E.; El-Ansari, R.; Craze, M.L.; Rakha, E.A.; Fan, S.-J.; Valli, A.; Haider, S.; Goberdhan, D.C.I.; Green, A.R.; et al. Increased expression of glutamine transporter SNAT2/SLC38A2 promotes glutamine dependence and oxidative stress resistance, and is associated with worse prognosis in triple-negative breast cancer. Br. J. Cancer 2021, 124, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, D.T.; McEwan, G.T.; Hirst, B.H.; Simmons, N.L. H(+)-coupled alpha-methylaminoisobutyric acid transport in human intestinal Caco-2 cells. Biochim. Biophys. Acta 1995, 1234, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fei, Y.J.; Anderson, C.M.; Wake, K.A.; Miyauchi, S.; Huang, W.; Thwaites, D.T.; Ganapathy, V. Structure, function and immunolocalization of a proton-coupled amino acid transporter (hPAT1) in the human intestinal cell line Caco-2. J. Physiol. 2003, 546, 349–361. [Google Scholar] [CrossRef]
- Nishimura, T.; Yagi, R.; Usuda, M.; Oda, K.; Yamazaki, M.; Suda, S.; Takahashi, Y.; Okazaki, F.; Sai, Y.; Higuchi, K.; et al. System A amino acid transporter SNAT2 shows subtype-specific affinity for betaine and hyperosmotic inducibility in placental trophoblasts. Biochim. Biophys. Acta 2014, 1838, 1306–1312. [Google Scholar] [CrossRef]
- Jakobsen, S.; Petersen, E.F.; Nielsen, C.U. Investigations of potential non-amino acid SNAT2 inhibitors. Front. Pharmacol. 2023, 14, 1302445. [Google Scholar] [CrossRef]
- Thwaites, D.T.; McEwan, G.T.; Cook, M.J.; Hirst, B.H.; Simmons, N.L. H(+)-coupled (Na(+)-independent) proline transport in human intestinal (Caco-2) epithelial cell monolayers. FEBS Lett. 1993, 333, 78–82. [Google Scholar] [CrossRef]
- Thwaites, D.T.; Basterfield, L.; McCleave, P.M.; Carter, S.M.; Simmons, N.L. Gamma-Aminobutyric acid (GABA) transport across human intestinal epithelial (Caco-2) cell monolayers. Br. J. Pharmacol. 2000, 129, 457–464. [Google Scholar] [CrossRef]
- Thwaites, D.T.; Stevens, B.C. H+-zwitterionic amino acid symport at the brush-border membrane of human intestinal epithelial (CACO-2) cells. Exp. Physiol. 1999, 84, 275–284. [Google Scholar]
- Thwaites, D.T.; McEwan, G.T.; Brown, C.D.; Hirst, B.H.; Simmons, N.L. L-alanine absorption in human intestinal Caco-2 cells driven by the proton electrochemical gradient. J. Membr. Biol. 1994, 140, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.; Holm, R.; Jensen, K.G.; Brodin, B.; Nielsen, C.U. Intestinal gaboxadol absorption via PAT1 (SLC36A1): Modified absorption in vivo following co-administration of L-tryptophan. Br. J. Pharmacol. 2009, 157, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.; Holm, R.; Jensen, K.G.; Sveigaard, C.; Brodin, B.; Nielsen, C.U. 5-Hydroxy-L-tryptophan alters gaboxadol pharmacokinetics in rats: Involvement of PAT1 and rOat1 in gaboxadol absorption and elimination. Eur. J. Pharm. Sci. 2010, 39, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Nøhr, M.K.; Hansen, S.H.; Brodin, B.; Holm, R.; Nielsen, C.U. The absorptive flux of the anti-epileptic drug substance vigabatrin is carrier-mediated across Caco-2 cell monolayers. Eur. J. Pharm. Sci. 2014, 51, 1–10. [Google Scholar] [CrossRef]
- Nøhr, M.K.; Thale, Z.I.; Brodin, B.; Hansen, S.H.; Holm, R.; Nielsen, C.U. Intestinal absorption of the antiepileptic drug substance vigabatrin is altered by infant formula in vitro and in vivo. Pharmacol. Res. Perspect. 2014, 2, e00036. [Google Scholar] [CrossRef]
- Nøhr, M.K.; Juul, R.V.; Thale, Z.I.; Holm, R.; Kreilgaard, M.; Nielsen, C.U. Is oral absorption of vigabatrin carrier-mediated? Eur. J. Pharm. Sci. 2015, 69, 10–18. [Google Scholar] [CrossRef]
- Broberg, M.; Holm, R.; Tønsberg, H.; Frølund, S.; Ewon, K.B.; Nielsen, A.; Brodin, B.; Jensen, A.; Kall, M.A.; Christensen, K.V.; et al. Function and expression of the proton-coupled amino acid transporter PAT1 along the rat gastrointestinal tract: Implications for intestinal absorption of gaboxadol. Br. J. Pharmacol. 2012, 167, 654–665. [Google Scholar] [CrossRef]
- Anderson, C.M.; Jevons, M.; Thangaraju, M.; Edwards, N.; Conlon, N.J.; Woods, S.; Ganapathy, V.; Thwaites, D.T. Transport of the photodynamic therapy agent 5-aminolevulinic acid by distinct H+-coupled nutrient carriers coexpressed in the small intestine. J. Pharmacol. Exp. Ther. 2010, 332, 220–228. [Google Scholar] [CrossRef]
- Abbot, E.L.; Grenade, D.S.; Kennedy, D.J.; Gatfield, K.M.; Thwaites, D.T. Vigabatrin transport across the human intestinal epithelial (Caco-2) brush-border membrane is via the H+ -coupled amino-acid transporter hPAT1. Br. J. Pharmacol. 2006, 147, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Frølund, S.; Marquez, O.C.; Larsen, M.; Brodin, B.; Nielsen, C.U. Delta-aminolevulinic acid is a substrate for the amino acid transporter SLC36A1 (hPAT1). Br. J. Pharmacol. 2010, 159, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Foltz, M.; Mertl, M.; Dietz, V.; Boll, M.; Kottra, G.; Daniel, H. Kinetics of bidirectional H+ and substrate transport by the proton-dependent amino acid symporter PAT1. Biochem. J. 2005, 386, 607–616. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vanslambrouck, J.M.; Bröer, A.; Thavyogarajah, T.; Holst, J.; Bailey, C.G.; Bröer, S.; Rasko, J.E. Renal imino acid and glycine transport system ontogeny and involvement in developmental iminoglycinuria. Biochem. J. 2010, 428, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Rostaing, P.; Ravassard, P.; Sagné, C.; Triller, A.; Giros, B. Lysosomal amino acid transporter LYAAT-1 in the rat central nervous system: An in situ hybridization and immunohistochemical study. J. Comp. Neurol. 2003, 462, 71–89. [Google Scholar] [CrossRef] [PubMed]
- Sagné, C.; Agulhon, C.; Ravassard, P.; Darmon, M.; Hamon, M.; El Mestikawy, S.; Gasnier, B.; Giros, B. Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc. Natl. Acad. Sci. USA 2001, 98, 7206–7211. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.; Figueiredo-Larsen, M.; Holm, R.; Broberg, M.L.; Brodin, B.; Nielsen, C.U. PAT1 (SLC36A1) shows nuclear localization and affects growth of smooth muscle cells from rats. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E65–E74. [Google Scholar] [CrossRef]
- The UniProt Consortium. SLC36A1—Proton-Coupled Amino Acid Transporter 1—Homo Sapiens (Human)|UniProtKB. Available online: https://www.uniprot.org/uniprotkb/Q7Z2H8/entry (accessed on 28 August 2023).
- Goberdhan, D.C.; Meredith, D.; Boyd, C.A.; Wilson, C. PAT-related amino acid transporters regulate growth via a novel mechanism that does not require bulk transport of amino acids. Development 2005, 132, 2365–2375. [Google Scholar] [CrossRef]
- Ögmundsdóttir, M.H.; Heublein, S.; Kazi, S.; Reynolds, B.; Visvalingam, S.M.; Shaw, M.K.; Goberdhan, D.C. Proton-assisted amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLoS ONE 2012, 7, e36616. [Google Scholar] [CrossRef]
- Schiöth, H.B.; Roshanbin, S.; Hägglund, M.G.; Fredriksson, R. Evolutionary origin of amino acid transporter families SLC32, SLC36 and SLC38 and physiological, pathological and therapeutic aspects. Mol. Aspects Med. 2013, 34, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Tuupanen, S.; Hänninen, U.A.; Kondelin, J.; von Nandelstadh, P.; Cajuso, T.; Gylfe, A.E.; Katainen, R.; Tanskanen, T.; Ristolainen, H.; Böhm, J.; et al. Identification of 33 candidate oncogenes by screening for base-specific mutations. Br. J. Cancer 2014, 111, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Donnard, E.; Asprino, P.F.; Correa, B.R.; Bettoni, F.; Koyama, F.C.; Navarro, F.C.; Perez, R.O.; Mariadason, J.; Sieber, O.M.; Strausberg, R.L.; et al. Mutational analysis of genes coding for cell surface proteins in colorectal cancer cell lines reveal novel altered pathways, druggable mutations and mutated epitopes for targeted therapy. Oncotarget 2014, 5, 9199–9213. [Google Scholar] [CrossRef] [PubMed]
- TCGA Research Network. SLC36A1—National Cancer Institute. Available online: https://portal.gdc.cancer.gov/genes/ENSG00000123643?canDistTable_size=100 (accessed on 28 August 2023).
- Luthra, S.; Chandran, U.; Diergaarde, B.; Becich, M.; Lee, A.V.; Neumann, C.A. Expression of reactive species related genes is associated with patient survival in luminal B breast cancer. Free Radic. Biol. Med. 2018, 120, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Feng, F.; Yang, Y.; Liu, K.; Duan, J.; Liu, H.; Yang, J.; Wu, M.; Liu, C.; Chang, Y. High-throughput screening identified miR-7-2-3p and miR-29c-3p as metastasis suppressors in gallbladder carcinoma. J. Gastroenterol. 2020, 55, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Metzner, L.; Kottra, G.; Neubert, K.; Daniel, H.; Brandsch, M. Serotonin, L-tryptophan, and tryptamine are effective inhibitors of the amino acid transport system PAT1. FASEB J. 2005, 19, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Frølund, S.; Langthaler, L.; Kall, M.A.; Holm, R.; Nielsen, C.U. Intestinal drug transport via the proton-coupled amino acid transporter PAT1 (SLC36A1) is inhibited by Gly-X(aa) dipeptides. Mol. Pharm. 2012, 9, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.U.; Frølund, S.; Abdulhadi, S.; Sari, H.; Langthaler, L.; Nøhr, M.K.; Kall, M.A.; Brodin, B.; Holm, R. Sertraline inhibits the transport of PAT1 substrates in vivo and in vitro. Br. J. Pharmacol. 2013, 170, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Yang, Y.; Wang, J.; Zuo, J.; Dong, X.; Li, C.; Li, D. Estradiol inhibits the activity of proton-coupled amino acid transporter PAT1 expressed in Xenopus oocytes. Eur. J. Pharmacol. 2012, 695, 34–39. [Google Scholar] [CrossRef]
- Nielsen, C.U.; Pedersen, M.; Müller, S.; Kæstel, T.; Bjerg, M.; Ulaganathan, N.; Nielsen, S.; Carlsen, K.L.; Nøhr, M.K.; Holm, R. Inhibitory Effects of 17-α-Ethinyl-Estradiol and 17-β-Estradiol on Transport Via the Intestinal Proton-Coupled Amino Acid Transporter (PAT1) Investigated In Vitro and In Vivo. J. Pharm. Sci. 2021, 110, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Nałęcz, K.A. Amino Acid Transporter SLC6A14 (ATB(0,+))—A Target in Combined Anti-cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 594464. [Google Scholar] [CrossRef]
- Karunakaran, S.; Ramachandran, S.; Coothankandaswamy, V.; Elangovan, S.; Babu, E.; Periyasamy-Thandavan, S.; Gurav, A.; Gnanaprakasam, J.P.; Singh, N.; Schoenlein, P.V.; et al. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J. Biol. Chem. 2011, 286, 31830–31838. [Google Scholar] [CrossRef]
- Coothankandaswamy, V.; Cao, S.; Xu, Y.; Prasad, P.D.; Singh, P.K.; Reynolds, C.P.; Yang, S.; Ogura, J.; Ganapathy, V.; Bhutia, Y.D. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br. J. Pharmacol. 2016, 173, 3292–3306. [Google Scholar] [CrossRef]
- Sniegowski, T.; Korac, K.; Bhutia, Y.D.; Ganapathy, V. SLC6A14 and SLC38A5 Drive the Glutaminolysis and Serine-Glycine-One-Carbon Pathways in Cancer. Pharmaceuticals 2021, 14, 126. [Google Scholar] [CrossRef]
- You, S.; Han, X.; Xu, Y.; Yao, Q. Research progress on the role of cationic amino acid transporter (CAT) family members in malignant tumors and immune microenvironment. Amino Acids 2023, 55, 1213–1222. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Cryo-EM enters a new era. eLife 2014, 3, e03678. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Rinaldi, S.; Scalbert, A.; Ferrari, P.; Achaintre, D.; Gunter, M.J.; Appleby, P.N.; Key, T.J.; Travis, R.C. Plasma concentrations and intakes of amino acids in male meat-eaters, fish-eaters, vegetarians and vegans: A cross-sectional analysis in the EPIC-Oxford cohort. Eur. J. Clin. Nutr. 2016, 70, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Console, L.; Galluccio, M.; Pochini, L.; Tonazzi, A.; Giangregorio, N.; Indiveri, C. Exploiting Cysteine Residues of SLC Membrane Transporters as Targets for Drugs. SLAS Discov. 2019, 24, 867–881. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | PDB | Conformation | Resolution, Å | Ligand |
|---|---|---|---|---|
| ASCT2 | 6GCT | IF (occluded) | 3.85 | L-Gln |
| 6MP6 | OF (open) | 3.54 | ||
| 6MPB | OF (occluded) | 3.84 | L-Gln | |
| 6RVX | IF (open) | 3.61 | ||
| 6RVY | IF (open) | 4.13 | ||
| 7BCQ | OF (open) | 3.43 | Lc-BPE (position “up”) | |
| 7BCS | OF (open) | 3.43 | Lc-BPE (position “down”) | |
| 7BCT | OF (open) | 3.37 | ||
| LAT1-4F2hc | 6IRS | IF (open) | 3.30 | |
| 6IRT | IF (open) | 3.50 | BCH | |
| 6JMQ | IF (open) | 3.31 | ||
| 7DSK | OF (occluded) | 2.90 | JX-075 | |
| 7DSL | OF (occluded) | 2.90 | JX-078 | |
| 7DSN | OF (occluded) | 3.10 | JX-119 | |
| 7DSQ | OF (open/occluded) | 3.40 | 3,5-diiodo-L-tyrosine | |
| xCT-4F2hc | 7CCS | IF (open) | 6.20 | |
| 7EPZ | IF (open) | 3.40 | Erastin | |
| 7P9U | IF (open) | 3.70 | L-Glu | |
| 7P9V | IF (open) | 3.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakobsen, S.; Nielsen, C.U. Exploring Amino Acid Transporters as Therapeutic Targets for Cancer: An Examination of Inhibitor Structures, Selectivity Issues, and Discovery Approaches. Pharmaceutics 2024, 16, 197. https://doi.org/10.3390/pharmaceutics16020197
Jakobsen S, Nielsen CU. Exploring Amino Acid Transporters as Therapeutic Targets for Cancer: An Examination of Inhibitor Structures, Selectivity Issues, and Discovery Approaches. Pharmaceutics. 2024; 16(2):197. https://doi.org/10.3390/pharmaceutics16020197
Chicago/Turabian StyleJakobsen, Sebastian, and Carsten Uhd Nielsen. 2024. "Exploring Amino Acid Transporters as Therapeutic Targets for Cancer: An Examination of Inhibitor Structures, Selectivity Issues, and Discovery Approaches" Pharmaceutics 16, no. 2: 197. https://doi.org/10.3390/pharmaceutics16020197
APA StyleJakobsen, S., & Nielsen, C. U. (2024). Exploring Amino Acid Transporters as Therapeutic Targets for Cancer: An Examination of Inhibitor Structures, Selectivity Issues, and Discovery Approaches. Pharmaceutics, 16(2), 197. https://doi.org/10.3390/pharmaceutics16020197