
A System for Discovering Novel Uricosurics Targeting Urate Transporter 1 Based on In Vitro and In Vivo Modeling

 , and
, and 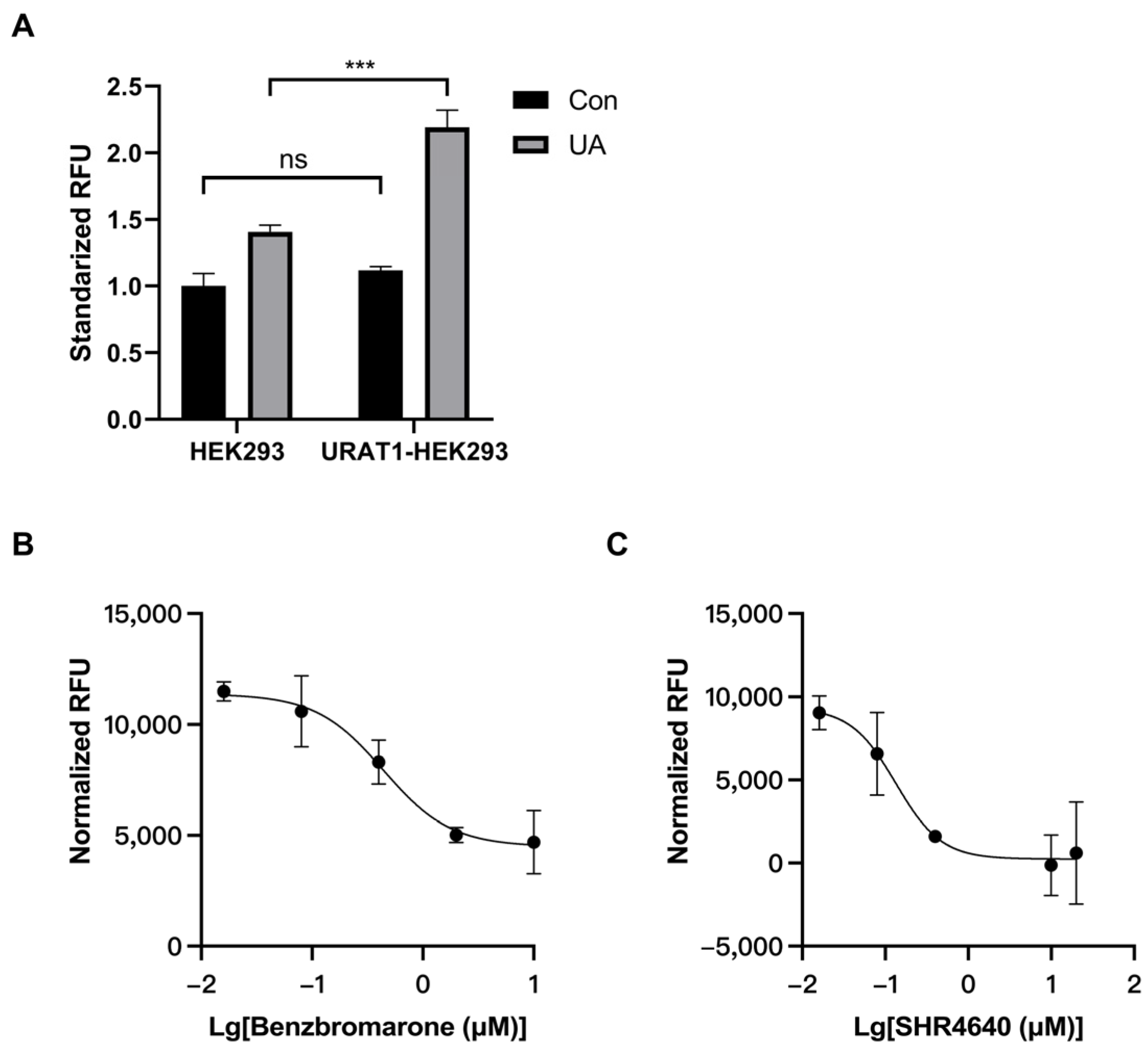{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Chemicals and Reagents
2.2. Animals
2.3. Cell Culture
2.4. Uric Acid Uptake Assay
2.5. Hyperuricemic Mice Models
2.6. Determination of Biochemical Parameters
2.7. Evaluation of XOR Activity In Vitro
2.8. Statistical Analysis
3. Results
3.1. Establishment of an In Vitro Model for URAT1 Inhibitor Preliminary Screening
3.2. Establishing the Hyperuricemic Mouse Models for Pharmacological Evaluation
3.3. Discovery of URAT1 Inhibitory Compound In Vitro
3.4. The Uric Acid-Lowering Effect of CC18002 In Vivo
3.5. CC18002 Had No Effect on XOR
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pascual, E.; Addadi, L.; Andrés, M.; Sivera, F. Mechanisms of crystal formation in gout—A structural approach. Nat. Rev. Rheumatol. 2015, 11, 725–730. [Google Scholar] [CrossRef]
- Ciarla, S.; Struglia, M.; Giorgini, P.; Striuli, R.; Necozione, S.; Properzi, G.; Ferri, C. Serum uric acid levels and metabolic syndrome. Arch. Physiol. Biochem. 2014, 120, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Feig, D.I.; Kang, D.H.; Johnson, R.J. Uric acid and cardiovascular risk. N. Engl. J. Med. 2008, 359, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Jalal, D.I.; Chonchol, M.; Chen, W.; Targher, G. Uric acid as a target of therapy in CKD. Am. J. Kidney Dis. 2013, 61, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Jalal, D.; Sanchez-Lozada, L.G.; Kang, D.H.; Ritz, E. Uric acid and chronic kidney disease: Which is chasing which? Nephrol. Dial. Transpl. 2013, 28, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Chen-Xu, M.; Yokose, C.; Rai, S.K.; Pillinger, M.H.; Choi, H.K. Contemporary prevalence of gout and hyperuricemia in the united states and decadal trends: The national health and nutrition examination survey, 2007–2016. Arthritis Rheumatol. 2019, 71, 991–999. [Google Scholar] [CrossRef]
- Smith, E.; March, L. Global prevalence of hyperuricemia: A systematic review of population-based epidemiological studies. Arthritis Rheumatol. 2015, 67, 10. [Google Scholar]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef]
- Fitzgerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American college of rheumatology guideline for the management of gout. Arthritis Care Res. 2020, 72, 744–760. [Google Scholar] [CrossRef]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef]
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Terkeltaub, R.; Bushinsky, D.A.; Becker, M.A. Recent developments in our understanding of the renal basis of hyperuricemia and the development of novel antihyperuricemic therapeutics. Arthritis Res. Ther. 2006, 8 (Suppl. S1), S4. [Google Scholar] [CrossRef][Green Version]
- Perez-Ruiz, F.; Alonso-Ruiz, A.; Calabozo, M.; Herrero-Beites, A.; Garcia-Erauskin, G.; Ruiz-Lucea, E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann. Rheum. Dis. 1998, 57, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Levinson, D.J.; Sorensen, L.B. Renal handling of uric acid in normal and gouty subject: Evidence for a 4-component system. Ann. Rheum. Dis. 1980, 39, 173–179. [Google Scholar] [CrossRef]
- Ndrepepa, G. Uric acid and cardiovascular disease. Clin. Chim. Acta 2018, 484, 150–163. [Google Scholar] [CrossRef]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Paredez, B.; Macias-Kauffer, L.; Fernandez-Lopez, J.C.; Villalobos-Comparan, M.; Martinez-Aguilar, M.M.; de la Cruz-Montoya, A.; Ramirez-Salazar, E.G.; Villamil-Ramirez, H.; Quiterio, M.; Ramirez-Palacios, P.; et al. Influence of genetic and Non-Genetic risk factors for serum uric acid levels and hyperuricemia in mexicans. Nutrients 2019, 11, 1336. [Google Scholar] [CrossRef]
- Hosoyamada, M.; Ichida, K.; Enomoto, A.; Hosoya, T.; Endou, H. Function and localization of urate transporter 1 in mouse kidney. J. Am. Soc. Nephrol. 2004, 15, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Hosoyamada, M.; Takiue, Y.; Morisaki, H.; Cheng, J.; Ikawa, M.; Okabe, M.; Morisaki, T.; Ichida, K.; Hosoya, T.; Shibasaki, T. Establishment and analysis of SLC22A12 (URAT1) knockout mouse. Nucleosides Nucleotides Nucleic Acids 2010, 29, 314–320. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The organic anion transporter (OAT) family: A systems biology perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef] [PubMed]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef]
- Yu, Z.; Fong, W.P.; Cheng, C.H. Morin (3,5,7,2′,4′-pentahydroxyflavone) exhibits potent inhibitory actions on urate transport by the human urate anion transporter (hURAT1) expressed in human embryonic kidney cells. Drug Metab. Dispos. 2007, 35, 981–986. [Google Scholar] [CrossRef]
- Azevedo, V.F.; Kos, I.A.; Vargas-Santos, A.B.; Pinheiro, G.d.R.C.; Paiva, E.d.S. Benzbromarone in the treatment of gout. Adv. Rheumatol. 2019, 59, 37. [Google Scholar] [CrossRef]
- Melethil, S.; Conway, W.D. Urinary excretion of probenecid and its metabolites in humans as a function of dose. J. Pharm. Sci. 1976, 65, 861–865. [Google Scholar] [CrossRef]
- Tan, P.K.; Ostertag, T.M.; Miner, J.N. Mechanism of high affinity inhibition of the human urate transporter URAT1. Sci. Rep. 2016, 6, 34995. [Google Scholar] [CrossRef] [PubMed]
- Strilchuk, L.; Fogacci, F.; Cicero, A.F. Safety and tolerability of available urate-lowering drugs: A critical review. Expert Opin. Drug Saf. 2019, 18, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Iqbal, K.; Farid, E.; Ishaque, A.; Hasanain, M.; Bin, A.T.; Arshad, A.S.; Rathore, S.S.; Malik, M. Efficacy and safety of dotinurad in hyperuricemic patients with or without gout: A systematic review and Meta-Analysis of randomized controlled trials. Cureus 2021, 13, e14428. [Google Scholar] [CrossRef]
- Peng, J.; Hu, Q.; Gu, C.; Liu, B.; Jin, F.; Yuan, J.; Feng, J.; Zhang, L.; Lan, J.; Dong, Q.; et al. Discovery of potent and orally bioavailable inhibitors of Human Uric Acid Transporter 1 (hURAT1) and binding mode prediction using homology model. Bioorg. Med. Chem. Lett. 2016, 26, 277–282. [Google Scholar] [CrossRef]
- Li, X.; Yan, Z.; Tian, J.; Zhang, X.; Han, H.; Ye, F. Urate transporter URAT1 in hyperuricemia: New insights from hyperuricemic models. Ann. Clin. Lab. Sci. 2019, 49, 756–762. [Google Scholar] [PubMed]
- Tan, P.K.; Liu, S.; Gunic, E.; Miner, J.N. Discovery and characterization of verinurad, a potent and specific inhibitor of URAT1 for the treatment of hyperuricemia and gout. Sci. Rep. 2017, 7, 665. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Chen, J.; Dong, S.; Li, H.; Cao, Y.; Tian, Y.; Fu, W.; Zhou, P.; Xi, B.; Pang, J. Identification and characterization of a potent and selective inhibitor of human urate transporter 1. Pharmacol. Rep. 2017, 69, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Zhou, X.; Liu, T.; Zhang, S.; Yuan, F.; Zhang, L.; Yang, Z.; Chen, Y. Astaxanthin attenuated hyperuricemia and kidney inflammation by inhibiting uric acid synthesis and the NF-kappa B/NLRP3 signaling pathways in potassium oxonate and hypoxanthine-induced hyperuricemia mice. Pharmazie 2021, 76, 551–558. [Google Scholar]
- Lin, C.; Zheng, Q.; Li, Y.; Wu, T.; Luo, J.; Jiang, Y.; Huang, Q.; Yan, C.; Zhang, L.; Zhang, W.; et al. Assessment of the influence on left ventricle by potassium oxonate and hypoxanthine-induced chronic hyperuricemia. Exp. Biol. Med. 2023, 248, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Yu, Q.; Xu, L.; Huang, Z.; Mai, L.; Jiang, L.; Su, Z.; Xie, J.; Li, Y.; Liu, Y.; et al. Berberrubine attenuates potassium oxonate- and hypoxanthine-induced hyperuricemia by regulating urate transporters and JAK2/STAT3 signaling pathway. Eur. J. Pharmacol. 2021, 912, 174592. [Google Scholar] [CrossRef]
- Chen, J.; Xu, L.; Jiang, L.; Wu, Y.; Wei, L.; Wu, X.; Xiao, S.; Liu, Y.; Gao, C.; Cai, J.; et al. Sonneratia apetala seed oil attenuates potassium oxonate/hypoxanthine-induced hyperuricemia and renal injury in mice. Food Funct. 2021, 12, 9416–9431. [Google Scholar] [CrossRef]
- Li, Q.; Huang, Z.; Liu, D.; Zheng, J.; Xie, J.; Chen, J.; Zeng, H.; Su, Z.; Li, Y. Effect of berberine on hyperuricemia and kidney injury: A network pharmacology analysis and experimental validation in a mouse model. Drug Des. Devel. Ther. 2021, 15, 3241–3254. [Google Scholar] [CrossRef]
- Yun, Y.; Yin, H.; Gao, Z.; Li, Y.; Gao, T.; Duan, J.; Yang, R.; Dong, X.; Zhang, L.; Duan, W. Intestinal tract is an important organ for lowering serum uric acid in rats. PLoS ONE 2017, 12, e190194. [Google Scholar] [CrossRef]
- Nishizawa, K.; Yoda, N.; Morokado, F.; Komori, H.; Nakanishi, T.; Tamai, I. Changes of drug pharmacokinetics mediated by downregulation of kidney organic cation transporters Mate1 and Oct2 in a rat model of hyperuricemia. PLoS ONE 2019, 14, e214862. [Google Scholar] [CrossRef]
- Preitner, F.; Pimentel, A.; Metref, S.; Berthonneche, C.; Sarre, A.; Moret, C.; Rotman, S.; Centeno, G.; Firsov, D.; Thorens, B. No development of hypertension in the hyperuricemic liver-Glut9 knockout mouse. Kidney Int. 2015, 87, 940–947. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Qi, C.; Shao, M.; Yang, Y.; Wang, Y.; Li, J.; Xiao, Z.; Ye, F. A System for Discovering Novel Uricosurics Targeting Urate Transporter 1 Based on In Vitro and In Vivo Modeling. Pharmaceutics 2024, 16, 172. https://doi.org/10.3390/pharmaceutics16020172
Li X, Qi C, Shao M, Yang Y, Wang Y, Li J, Xiao Z, Ye F. A System for Discovering Novel Uricosurics Targeting Urate Transporter 1 Based on In Vitro and In Vivo Modeling. Pharmaceutics. 2024; 16(2):172. https://doi.org/10.3390/pharmaceutics16020172
Chicago/Turabian StyleLi, Xuechen, Chufan Qi, Mengjie Shao, Yajun Yang, Yuying Wang, Jiang Li, Zhiyan Xiao, and Fei Ye. 2024. "A System for Discovering Novel Uricosurics Targeting Urate Transporter 1 Based on In Vitro and In Vivo Modeling" Pharmaceutics 16, no. 2: 172. https://doi.org/10.3390/pharmaceutics16020172
APA StyleLi, X., Qi, C., Shao, M., Yang, Y., Wang, Y., Li, J., Xiao, Z., & Ye, F. (2024). A System for Discovering Novel Uricosurics Targeting Urate Transporter 1 Based on In Vitro and In Vivo Modeling. Pharmaceutics, 16(2), 172. https://doi.org/10.3390/pharmaceutics16020172