
Modified PEG-Lipids Enhance the Nasal Mucosal Immune Capacity of Lipid Nanoparticle mRNA Vaccines

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
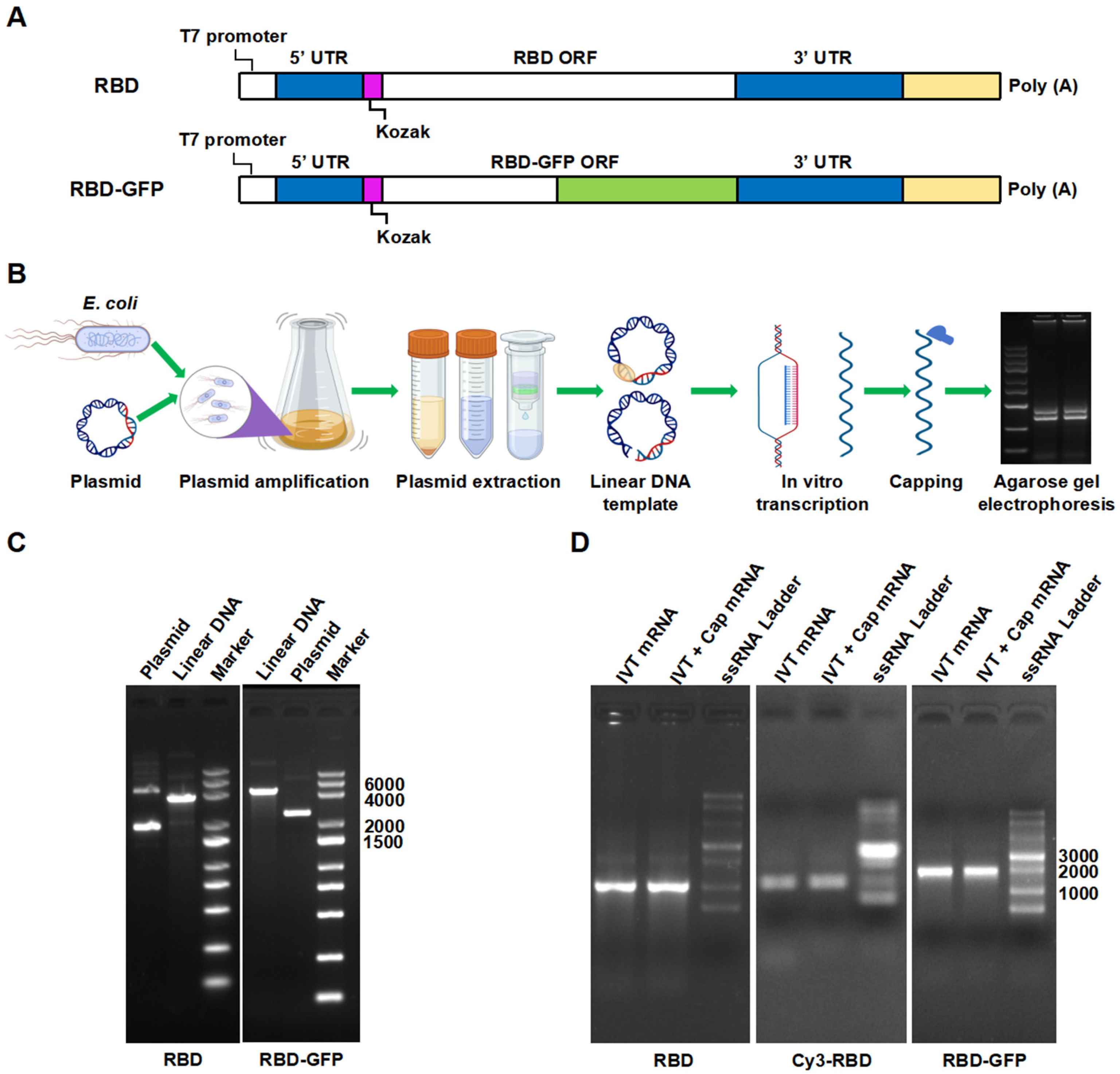
2.2.1. mRNA Preparation and Identification
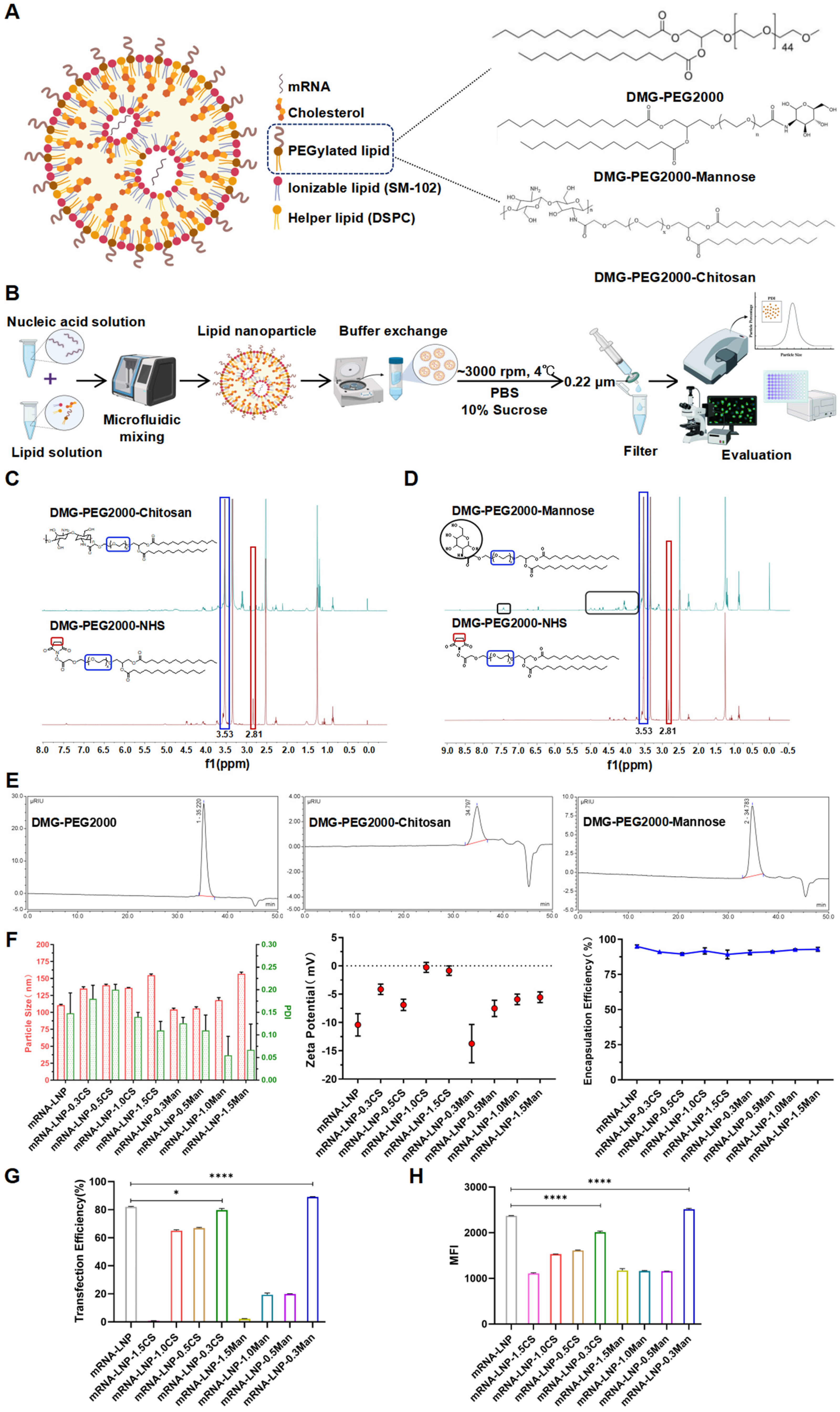
2.2.2. LNP Formulation and Characterization
2.2.3. Transfection Efficiency of LNPs in DC2.4 Cells
2.2.4. Morphological Characterization of LNPs Using Transmission Electron Microscopy (TEM)
2.2.5. Cellular Uptake
2.2.6. Endosome Escape
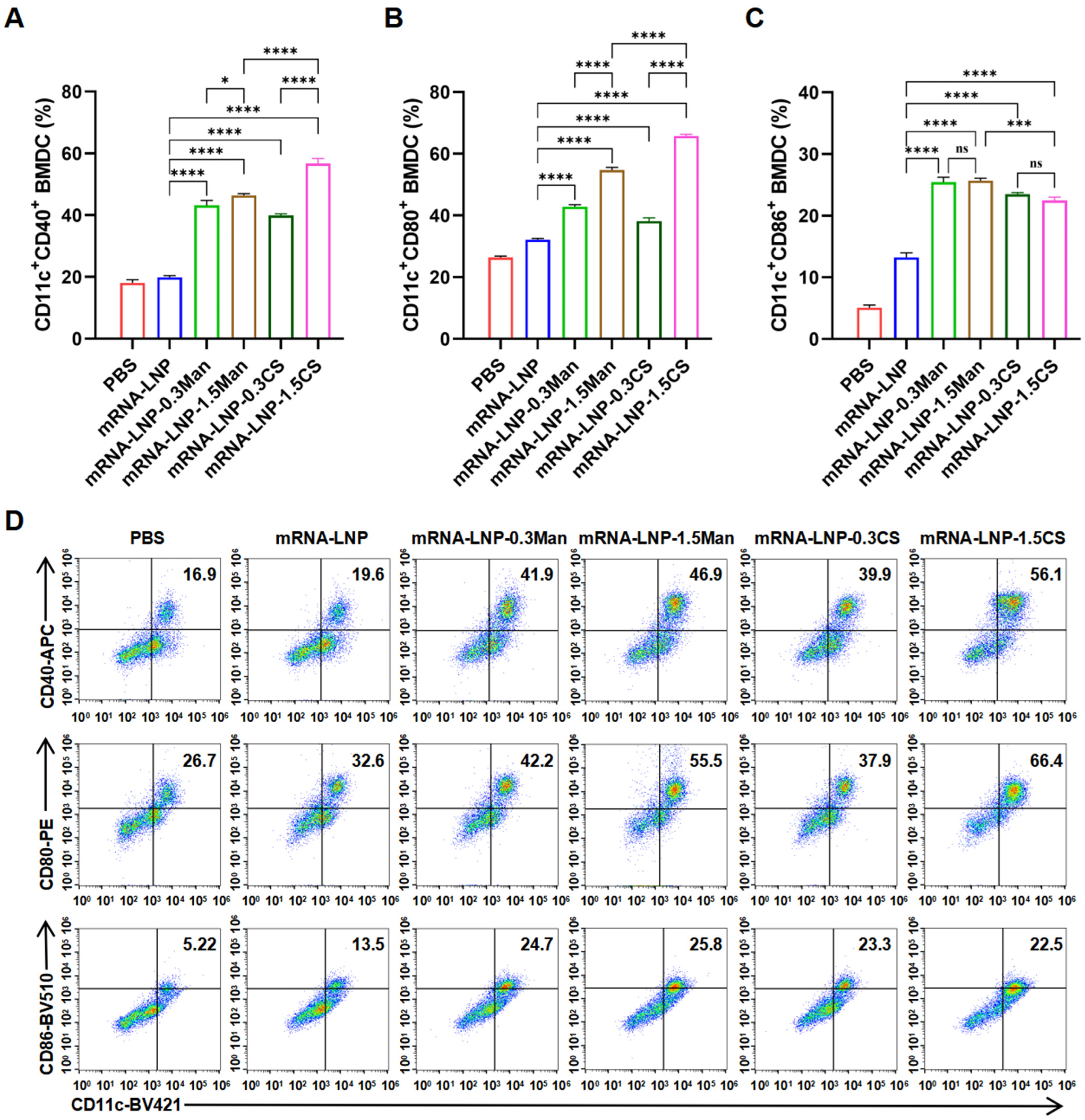
2.2.7. BMDCs Activation Experiment
2.2.8. Immunization
2.2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
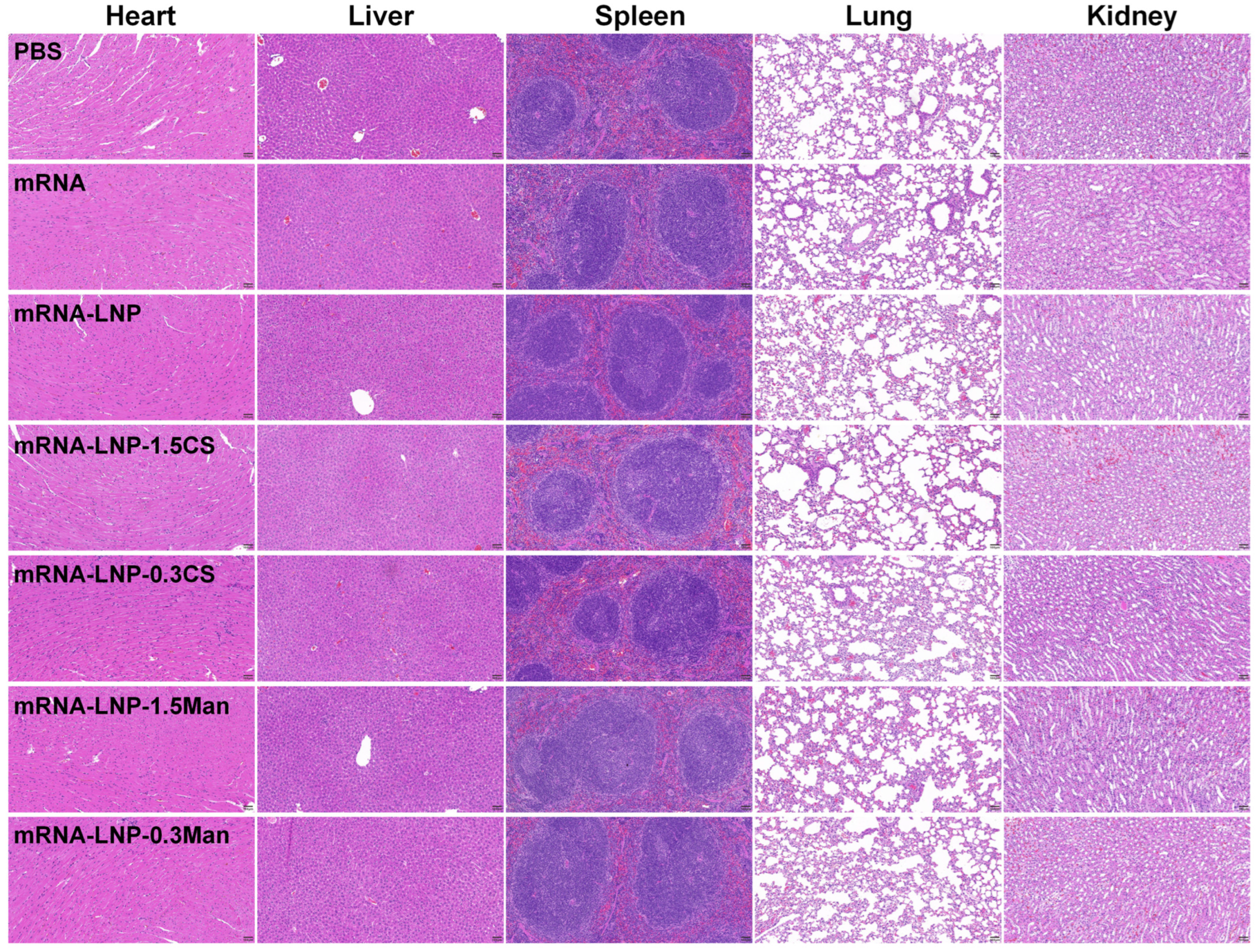
2.2.10. Hematoxylin and Eosin (H&E) Staining
2.2.11. Statistical Analysis
3. Results and Discussion
3.1. mRNA Identification
3.2. LNP Formulation and Characterization
3.3. Transfection Efficiency of LNPs in DC2.4 Cells
3.4. Morphological Characterization of LNPs Using TEM
3.5. Cellular Uptake
3.6. Endosome Escape
3.7. BMDCs Activation Experiment
3.8. Immunization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Diallo, B.K.; Ní Chasaide, C.; Wong, T.Y.; Schmitt, P.; Lee, K.S.; Weaver, K.; Miller, O.; Cooper, M.; Jazayeri, S.D.; Damron, F.H.; et al. Intranasal COVID-19 vaccine induces respiratory memory T cells and protects K18-hACE mice against SARS-CoV-2 infection. NPJ Vaccines 2023, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Zhang, W. SARS-CoV-2 variants, immune escape, and countermeasures. Front. Med. 2022, 16, 196–207. [Google Scholar] [CrossRef]
- Ssentongo, P.; Ssentongo, A.E.; Voleti, N.; Groff, D.; Sun, A.; Ba, D.M.; Nunez, J.; Parent, L.J.; Chinchilli, V.M.; Paules, C.I. SARS-CoV-2 vaccine effectiveness against infection, symptomatic and severe COVID-19: A systematic review and meta-analysis. BMC Infect. Dis. 2022, 22, 439. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug. Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Azzi, L.; Dalla Gasperina, D.; Veronesi, G.; Shallak, M.; Ietto, G.; Iovino, D.; Baj, A.; Gianfagna, F.; Maurino, V.; Focosi, D.; et al. Mucosal immune response in BNT162b2 COVID-19 vaccine recipients. eBioMedicine 2022, 75, 103788. [Google Scholar] [CrossRef]
- Tang, J.; Zeng, C.; Cox, T.M.; Li, C.; Son, Y.M.; Cheon, I.S.; Wu, Y.; Behl, S.; Taylor, J.J.; Chakaraborty, R.; et al. Respiratory mucosal immunity against SARS-CoV-2 after mRNA vaccination. Sci. Immunol. 2022, 7, eadd4853. [Google Scholar] [CrossRef]
- Altarawneh, H.N.; Chemaitelly, H.; Ayoub, H.H.; Hasan, M.R.; Coyle, P.; Yassine, H.M.; Al-Khatib, H.A.; Smatti, M.K.; Al-Kanaani, Z.; Al-Kuwari, E.; et al. Protective effect of previous SARS-CoV-2 infection against omicron BA.4 and BA.5 subvariants. N. Engl. J. Med. 2022, 387, 1620–1622. [Google Scholar] [CrossRef]
- Marks, M.; Millat-Martinez, P.; Ouchi, D.; Roberts, C.h.; Alemany, A.; Corbacho-Monné, M.; Ubals, M.; Tobias, A.; Tebé, C.; Ballana, E.; et al. Transmission of COVID-19 in 282 clusters in Catalonia, Spain: A cohort study. Lancet Infect. Dis. 2021, 21, 629–636. [Google Scholar] [CrossRef]
- Liew, F.; Talwar, S.; Cross, A.; Willett, B.J.; Scott, S.; Logan, N.; Siggins, M.K.; Swieboda, D.; Sidhu, J.K.; Efstathiou, C.; et al. SARS-CoV-2-specific nasal IgA wanes 9 months after hospitalisation with COVID-19 and is not induced by subsequent vaccination. eBioMedicine 2023, 87, 104402. [Google Scholar] [CrossRef]
- Wong, T.Y.; Lee, K.S.; Russ, B.P.; Horspool, A.M.; Kang, J.; Winters, M.T.; Allison Wolf, M.; Rader, N.A.; Miller, O.A.; Shiflett, M.; et al. Intranasal administration of BReC-CoV-2 COVID-19 vaccine protects K18-hACE2 mice against lethal SARS-CoV-2 challenge. NPJ Vaccines 2022, 7, 36. [Google Scholar] [CrossRef]
- Alu, A.; Chen, L.; Lei, H.; Wei, Y.; Tian, X.; Wei, X. Intranasal COVID-19 vaccines: From bench to bed. eBioMedicine 2022, 76, 103841. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Xu, Y.; Feng, J.; Hu, L.; Zhang, Y.; Zhang, B.; Guo, W.; Mai, R.; Chen, L.; Fang, J.; et al. Intranasal administration of a recombinant RBD vaccine induced protective immunity against SARS-CoV-2 in mouse. Vaccine 2021, 39, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, M.; Fu, Y.; Li, Y.; Gong, T.; Zhang, Z.; Sun, X. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways. J. Control. Release 2016, 228, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines 2014, 14, 161–176. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef]
- Schattling, P.; Taipaleenmäki, E.; Zhang, Y.; Städler, B. A Polymer Chemistry Point of View on Mucoadhesion and Mucopenetration. Macromol. Biosci. 2017, 17, 1700060. [Google Scholar] [CrossRef]
- Lang, X.; Wang, T.; Sun, M.; Chen, X.; Liu, Y. Advances and applications of chitosan-based nanomaterials as oral delivery carriers: A review. Int. J. Biol. Macromol. 2020, 154, 433–445. [Google Scholar] [CrossRef]
- Dumkliang, E.; Pamornpathomkul, B.; Patrojanasophon, P.; Ngawhirunpat, T.; Rojanarata, T.; Yoksan, S.; Opanasopit, P. Feasibility of chitosan-based nanoparticles approach for intranasal immunisation of live attenuated Japanese encephalitis vaccine. Int. J. Biol. Macromol. 2021, 183, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Mosafer, J.; Sabbaghi, A.-H.; Badiee, A.; Dehghan, S.; Tafaghodi, M. Preparation, characterization and in vivo evaluation of alginate-coated chitosan and trimethylchitosan nanoparticles loaded with PR8 influenza virus for nasal immunization. Asian J. Pharm. Sci. 2019, 14, 216–221. [Google Scholar] [CrossRef]
- Lopes, P.D.; Okino, C.H.; Fernando, F.S.; Pavani, C.; Casagrande, V.M.; Lopez, R.F.V.; Montassier, M.d.F.S.; Montassier, H.J. Inactivated infectious bronchitis virus vaccine encapsulated in chitosan nanoparticles induces mucosal immune responses and effective protection against challenge. Vaccine 2018, 36, 2630–2636. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, S.; Haddadi, A.; Shayeganpour, A.; Samuel, J.; Lavasanifar, A. Activation of Antigen-Specific T Cell-Responses by Mannan-Decorated PLGA Nanoparticles. Pharm. Res. 2011, 28, 2288–2301. [Google Scholar] [CrossRef]
- Li, H.-S.; Shin, M.-K.; Singh, B.; Maharjan, S.; Park, T.-E.; Kang, S.-K.; Yoo, H.-S.; Hong, Z.-S.; Cho, C.-S.; Choi, Y.-J. Nasal immunization with mannan-decorated mucoadhesive HPMCP microspheres containing ApxIIA toxin induces protective immunity against challenge infection with Actinobacillus pleuropneumoiae in mice. J. Control. Release 2016, 233, 114–125. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, W.; Song, Y.; Wang, W.; Zhang, Y.; Wang, T.; Li, K.; Pan, Q.; Qi, X.; Gao, Y.; et al. Recombinant Lactococcus lactis co-expressing OmpH of an M cell-targeting ligand and IBDV-VP2 protein provide immunological protection in chickens. Vaccine 2018, 36, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, J.; Wang, J.; Zhang, W.; Xu, B.; Xu, X.; Zong, L. Targeted delivery of antigen to intestinal dendritic cells induces oral tolerance and prevents autoimmune diabetes in NOD mice. Diabetologia 2018, 61, 1384–1396. [Google Scholar] [CrossRef]
- Hald Albertsen, C.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, Y.; Hihara, T.; Kubara, K.; Kondo, K.; Hyodo, K.; Yamazaki, K.; Ishida, T.; Ishihara, H. PEG shedding-rate-dependent blood clearance of PEGylated lipid nanoparticles in mice: Faster PEG shedding attenuates anti-PEG IgM production. Int. J. Pharm. 2020, 588, 119792. [Google Scholar] [CrossRef]
- Judge, A.; McClintock, K.; Phelps, J.R.; MacLachlan, I. Hypersensitivity and Loss of Disease Site Targeting Caused by Antibody Responses to PEGylated Liposomes. Mol. Ther. 2006, 13, 328–337. [Google Scholar] [CrossRef]
- Mui, B.L.; Tam, Y.K.; Jayaraman, M.; Ansell, S.M.; Du, X.; Tam, Y.Y.C.; Lin, P.J.C.; Chen, S.; Narayanannair, J.K.; Rajeev, K.G.; et al. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles. Mol. Ther. Nucleic Acids 2013, 2, e139. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.C.; Harasym, T.O.; Clow, K.A.; Ansell, S.M.; Klimuk, S.K.; Hope, M.J. Immunogenicity and Rapid Blood Clearance of Liposomes Containing Polyethylene Glycol-Lipid Conjugates and Nucleic Acid. J. Pharmacol. Exp. Ther. 2005, 312, 1020–1026. [Google Scholar] [CrossRef]
- Bandekar, A.; Zarraga, I.E.; Dugas, J.; Ghosh, P.; Bailey-Hytholt, C.M. Formulating and Characterizing Lipid Nanoparticles for Gene Delivery using a Microfluidic Mixing Platform. J. Vis. Exp. 2021, 168, e62226. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Kaminskas, L.M.; Porter, C.J.H. From sewer to saviour—Targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug. Discov. 2015, 14, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Lahann, J. Physical approaches to biomaterial design. Nat. Mater. 2009, 8, 15–23. [Google Scholar] [CrossRef]
- Barua, S.; Mitragotri, S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef]
- Holland, J.W.; Hui, C.; Cullis, P.R.; Madden, T.D. Poly(ethylene glycol)—Lipid conjugates regulate the calcium-induced fusion of liposomes composed of phosphatidylethanolamine and phosphatidylserine. Biochemistry 1996, 35, 2618–2624. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, F.; Yanez Arteta, M.; Lerche, M.; Porcar, L.; Lang, C.; Bragg, R.A.; Elmore, C.S.; Krishnamurthy, V.R.; Russell, R.A.; Darwish, T.; et al. Apolipoprotein E Binding Drives Structural and Compositional Rearrangement of mRNA-Containing Lipid Nanoparticles. ACS Nano 2021, 15, 6709–6722. [Google Scholar] [CrossRef]
- Eygeris, Y.; Gupta, M.; Kim, J.; Sahay, G. Chemistry of Lipid Nanoparticles for RNA Delivery. Acc. Chem. Res. 2021, 55, 2–12. [Google Scholar] [CrossRef]
- Dutta, D.; Donaldson, J.G. Search for inhibitors of endocytosis. Cell. Logist. 2014, 2, 203–208. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Bio. 2011, 12, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Šamaj, J.; Baluška, F.; Voigt, B.; Schlicht, M.; Volkmann, D.; Menzel, D. Endocytosis, Actin Cytoskeleton, and Signaling. Plant Physiol. 2004, 135, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Gandek, T.B.; van der Koog, L.; Nagelkerke, A. A Comparison of Cellular Uptake Mechanisms, Delivery Efficacy, and Intracellular Fate between Liposomes and Extracellular Vesicles. Adv. Healthc. Mater. 2023, 12, e2300319. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, D.; Ceña, V. Endocytosis: The Nanoparticle and Submicron Nanocompounds Gateway into the Cell. Pharmaceutics 2020, 12, 371. [Google Scholar] [CrossRef]
- Sieczkarski, S.B.; Whittaker, G.R. Dissecting virus entry via endocytosis. J. Gen. Virol. 2002, 83, 1535–1545. [Google Scholar] [CrossRef]
- Francia, V.; Reker-Smit, C.; Boel, G.; Salvati, A. Limits and challenges in using transport inhibitors to characterize how nano-sized drug carriers enter cells. Nanomedicine 2019, 14, 1533–1549. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions About Dendritic Cells: Past, Present, and Future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef]
- Hopkins, R.A.; Connolly, J.E. The specialized roles of immature and mature dendritic cells in antigen cross-presentation. Immunol. Res. 2012, 53, 91–107. [Google Scholar] [CrossRef]
- Chen, M.-H.; Li, W.-S.; Lue, Y.-S.; Chu, C.-L.; Pan, I.H.; Ko, C.-H.; Chen, D.-Y.; Lin, C.-H.; Lin, S.-H.; Chang, C.-P.; et al. Clitocybe nudaActivates Dendritic Cells and Acts as a DNA Vaccine Adjuvant. Evid. Based Complement. Altern. Med. 2013, 2013, 761454. [Google Scholar] [CrossRef]
- Zughaier, S.; Agrawal, S.; Stephens, D.; Pulendran, B. Hexa-acylation and KDO2-glycosylation determine the specific immunostimulatory activity of Neisseria meningitidis lipid A for human monocyte derived dendritic cells. Vaccine 2006, 24, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liang, X.; Mao, C.; Jiang, Y. The Distinct Properties of Polysaccharide Nanoparticles Tune Immune Responses against mRNA Antigen via Stimulator of Interferon Genes-Mediated Autophagy and Inflammasome. ACS Nano 2023, 17, 21782–21798. [Google Scholar] [CrossRef]
- Gao, X.; Xiong, Y.; Chen, H.; Gao, X.; Dai, J.; Zhang, Y.; Zou, W.; Gao, Y.; Jiang, Z.; Han, B. Mucus adhesion vs. mucus penetration? Screening nanomaterials for nasal inhalation by MD simulation. J. Control. Release 2023, 353, 366–379. [Google Scholar] [CrossRef]
- Rostamian, M.; Sohrabi, S.; Kavosifard, H.; Niknam, H.M. Lower levels of IgG1 in comparison with IgG2a are associated with protective immunity against Leishmania tropica infection in BALB/c mice. J. Microbiol. Immunol. 2017, 50, 160–166. [Google Scholar] [CrossRef]
- Zhao, F.; Qu, J.; Wang, W.; Li, S.; Xu, S. The imbalance of Th1/Th2 triggers an inflammatory response in chicken spleens after ammonia exposure. Poultry Sci. 2020, 99, 3817–3822. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhou, M.; Yu, S.; Jin, Z.; Zhao, K. An overview of biodegradable nanomaterials and applications in vaccines. Vaccine 2020, 38, 1096–1104. [Google Scholar]
- Alwahsh, W.; Sahudin, S.; Alkhatib, H.; Bostanudin, M.F.; Alwahsh, M. Chitosan-Based Nanocarriers for Pulmonary and Intranasal Drug Delivery Systems: A Comprehensive Overview of their Applications. Curr. Drug Targets 2024, 25, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Virmani, T.; Kumar, G.; Sharma, A.; Pathak, K.; Akhtar, M.S.; Afzal, O.; Altamimi, A.S.A. Amelioration of Cancer Employing Chitosan, Its Derivatives, and Chitosan-Based Nanoparticles: Recent Updates. Polymers 2023, 15, 2928. [Google Scholar] [CrossRef]
- Şenel, S.; Yüksel, S. Chitosan-based particulate systems for drug and vaccine delivery in the treatment and prevention of neglected tropical diseases. Drug Deliv. Transl. Res. 2020, 10, 1644–1674. [Google Scholar] [CrossRef]
- Bugnicourt, L.; Ladavière, C. A close collaboration of chitosan with lipid colloidal carriers for drug delivery applications. J. Control. Release 2017, 256, 121–140. [Google Scholar] [CrossRef]
- Tezgel, Ö.; Szarpak-Jankowska, A.; Arnould, A.; Auzély-Velty, R.; Texier, I. Chitosan-lipid nanoparticles (CS-LNPs): Application to siRNA delivery. J. Colloid Interface Sci. 2018, 510, 45–56. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | SM-102 (mol%) | DSPC (mol%) | Cholesterol (mol%) | DMG-PEG2000 (mol%) | DMG-PEG2000-Chitosan (mol%) | DMG-PEG2000-Mannose (mol%) | N/P | Total Flow (mL/min) |
|---|---|---|---|---|---|---|---|---|
| mRNA-LNP | 50 | 10 | 38.5 | 1.5 | 0 | 0 | 6 | 8 |
| mRNA-LNP-1.5CS | 50 | 10 | 38.5 | 0 | 1.5 | 0 | 6 | 12 |
| mRNA-LNP-1.0CS | 50 | 10 | 38.5 | 0.5 | 1.0 | 0 | 6 | 12 |
| mRNA-LNP-0.5CS | 50 | 10 | 38.5 | 1.0 | 0.5 | 0 | 6 | 12 |
| mRNA-LNP-0.3CS | 50 | 10 | 38.5 | 1.2 | 0.3 | 0 | 6 | 12 |
| mRNA-LNP-1.5Man | 50 | 10 | 38.5 | 0 | 0 | 1.5 | 6 | 16 |
| mRNA-LNP-1.0Man | 50 | 10 | 38.5 | 0.5 | 0 | 1.0 | 6 | 16 |
| mRNA-LNP-0.5Man | 50 | 10 | 38.5 | 1.0 | 0 | 0.5 | 6 | 16 |
| mRNA-LNP-0.3Man | 50 | 10 | 38.5 | 1.2 | 0 | 0.3 | 6 | 16 |
| Inhibitor | Function | Concentration |
|---|---|---|
| Methyl-β-cyclodextrin (MβCD) | Inhibition of caveolae/lipid raft mediated endocytosis by cholesterol depletion | 10 μg/mL |
| Chlorpromazine | Inhibitor of clathrin | 10 μg/mL |
| Nystatin | Inhibitor of caveolae/lipid raft | 25 μg/mL |
| Amiloride (EIPA) | Inhibitor of macropinocytosis | 15 μg/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Yi, J.; Lu, Y.; Liu, T.; Xing, H.; Wang, X.; Zhang, H.; Liu, N.; Wang, Z.; Zheng, A. Modified PEG-Lipids Enhance the Nasal Mucosal Immune Capacity of Lipid Nanoparticle mRNA Vaccines. Pharmaceutics 2024, 16, 1423. https://doi.org/10.3390/pharmaceutics16111423
Li M, Yi J, Lu Y, Liu T, Xing H, Wang X, Zhang H, Liu N, Wang Z, Zheng A. Modified PEG-Lipids Enhance the Nasal Mucosal Immune Capacity of Lipid Nanoparticle mRNA Vaccines. Pharmaceutics. 2024; 16(11):1423. https://doi.org/10.3390/pharmaceutics16111423
Chicago/Turabian StyleLi, Meng, Jing Yi, Yicheng Lu, Ting Liu, Haonan Xing, Xiwei Wang, Hui Zhang, Nan Liu, Zengming Wang, and Aiping Zheng. 2024. "Modified PEG-Lipids Enhance the Nasal Mucosal Immune Capacity of Lipid Nanoparticle mRNA Vaccines" Pharmaceutics 16, no. 11: 1423. https://doi.org/10.3390/pharmaceutics16111423
APA StyleLi, M., Yi, J., Lu, Y., Liu, T., Xing, H., Wang, X., Zhang, H., Liu, N., Wang, Z., & Zheng, A. (2024). Modified PEG-Lipids Enhance the Nasal Mucosal Immune Capacity of Lipid Nanoparticle mRNA Vaccines. Pharmaceutics, 16(11), 1423. https://doi.org/10.3390/pharmaceutics16111423