

Maximizing Anticancer Response with MPS1 and CENPE Inhibition Alongside Apoptosis Induction


 ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Small Molecule Inhibitors
2.2. Cell Culture
2.3. RNA Extraction, cDNA Synthesis and Quantitative Real-Time PCR
2.4. Protein Extracts and Western Blotting
2.5. Indirect Immunofluorescence
2.6. MTT Viability Assay
2.7. Apoptosis Detection
2.7.1. TUNEL Assay
2.7.2. Annexin V/PI Staining
2.8. Mitotic Index Determination
2.9. Time-Lapse Microscopy
2.10. Phase-Contrast and Fluorescence Microscopy Images
2.11. Colony Formation Assay
2.12. Caspase Activity Assay
2.13. Spheroid Formation, Drug Treatment and Viability Assay
2.14. Statistical Analysis
3. Results
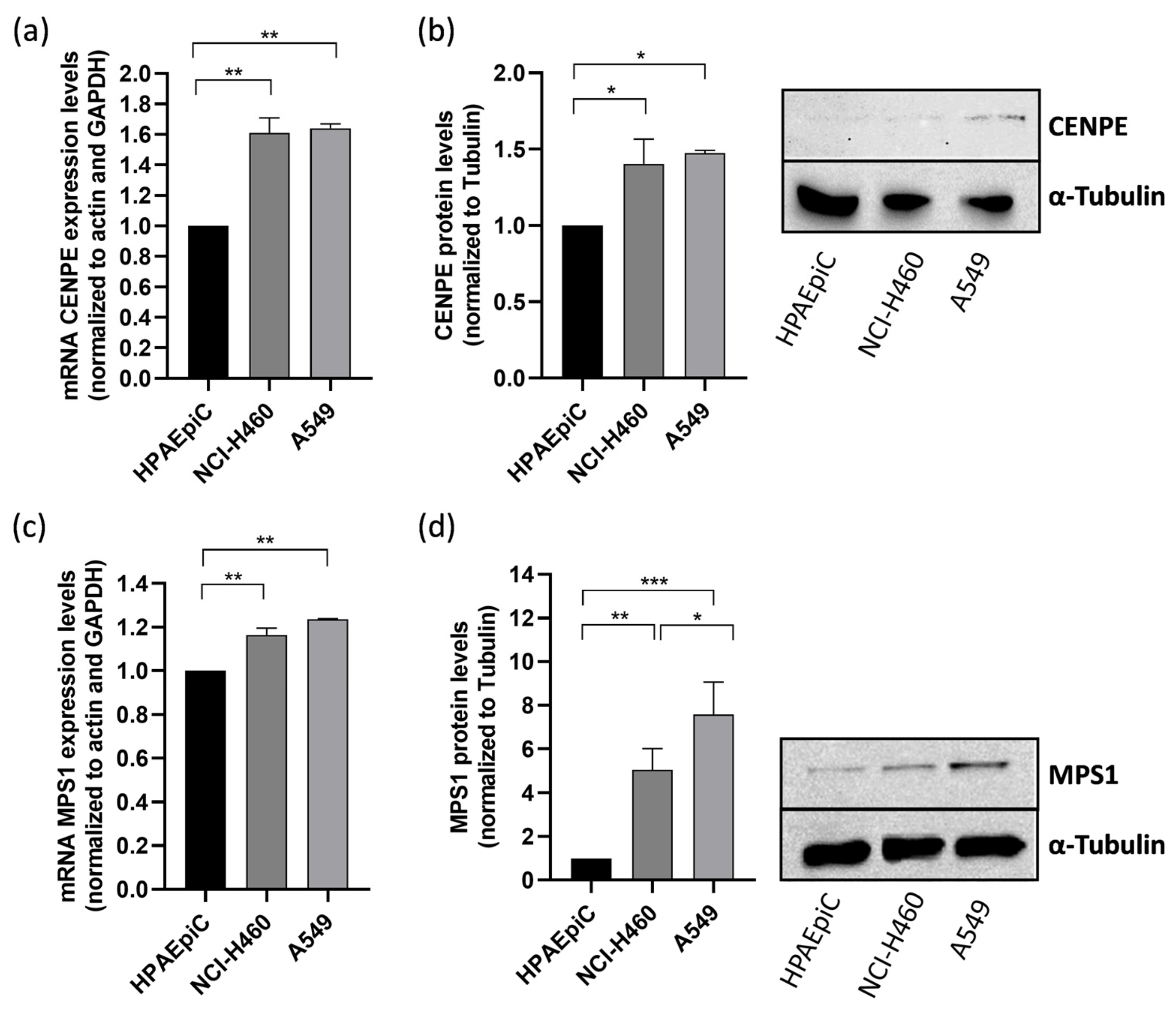
3.1. CENPE and MPS1 Are Overexpressed in Lung Cancer Cells
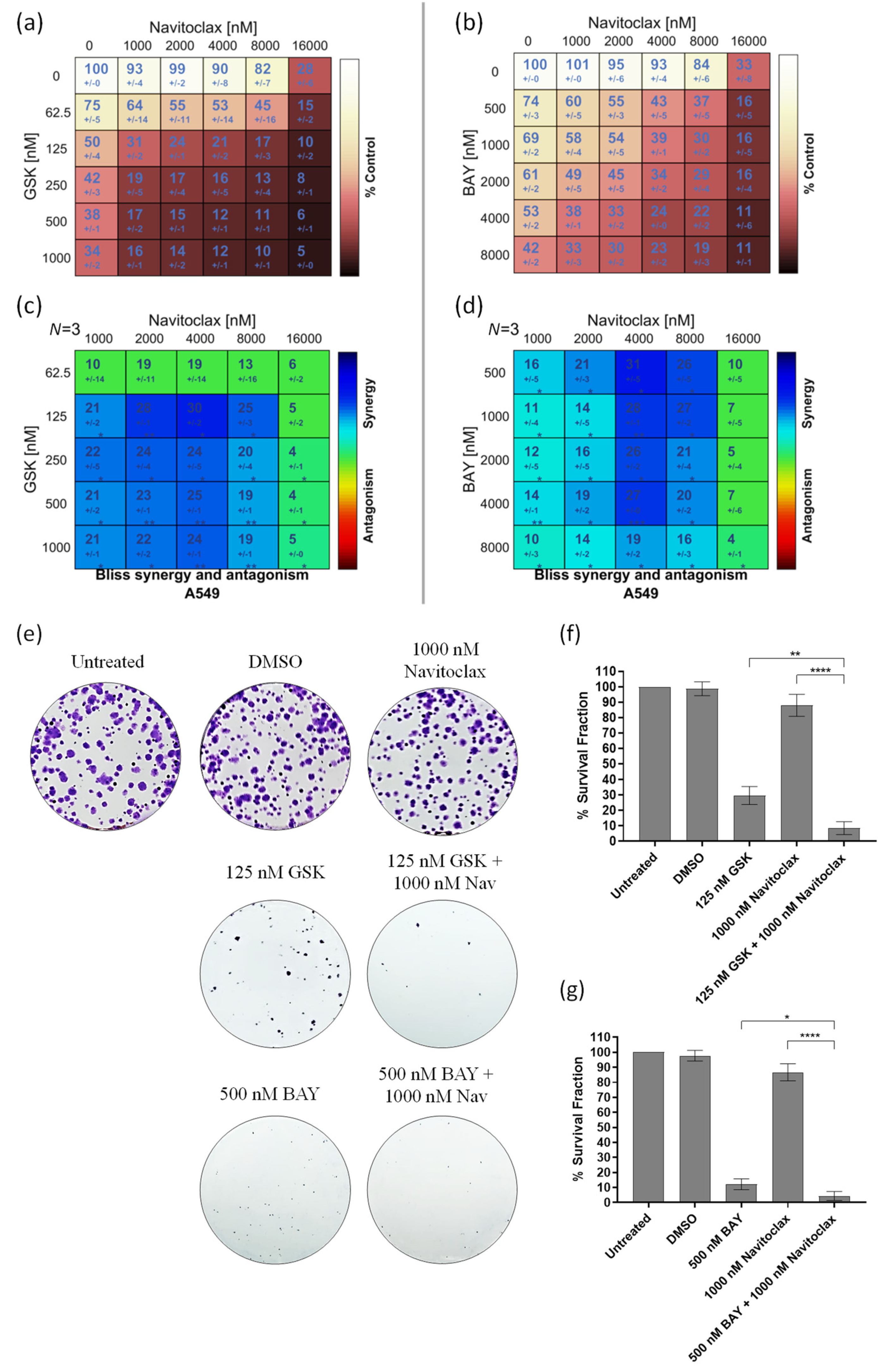
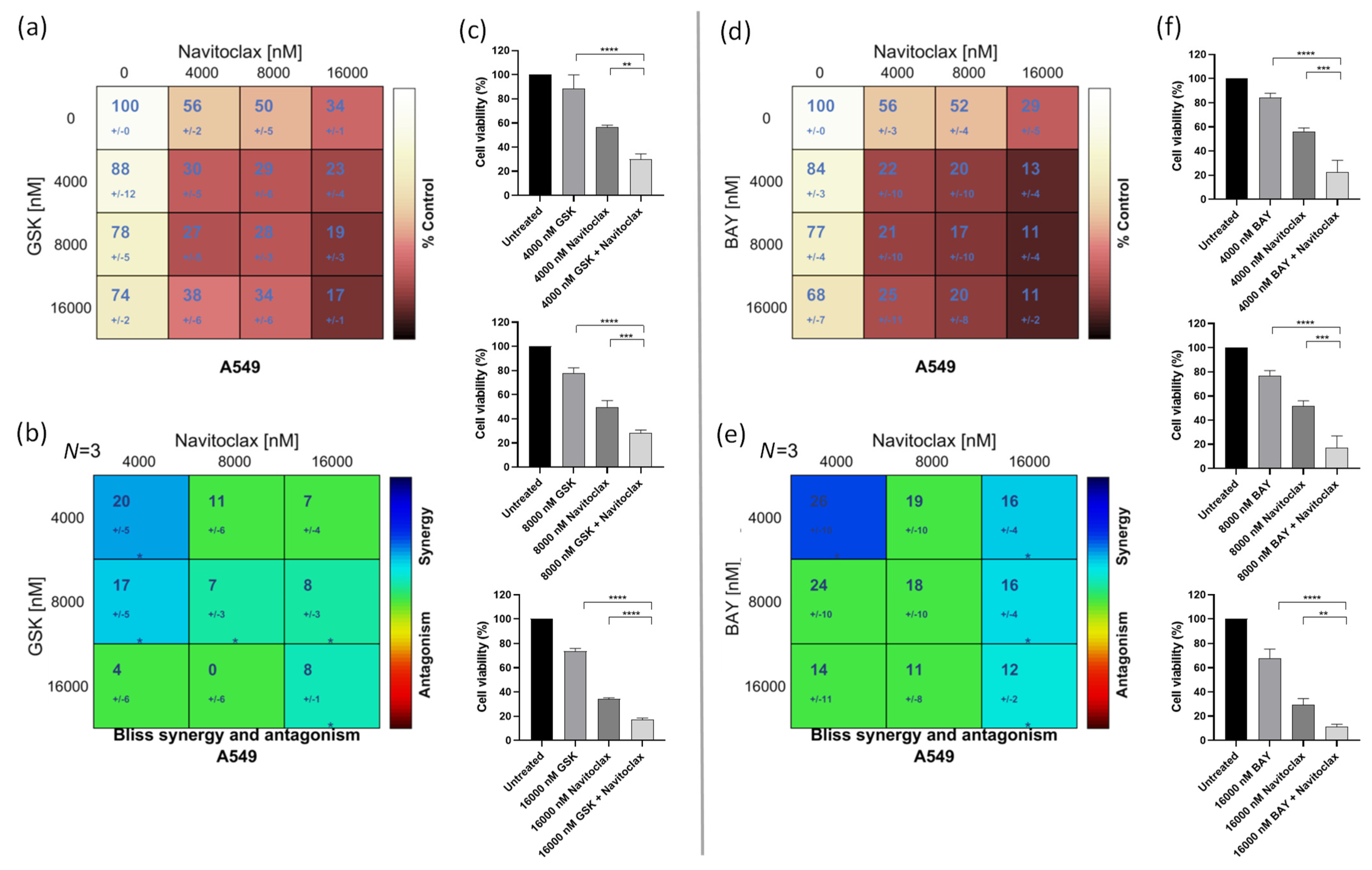
3.2. Navitoclax Synergizes with the Mitotic Blocker GSK923295 and the Mitotic Driver BAY1217389 in Killing Lung Cancer Cells
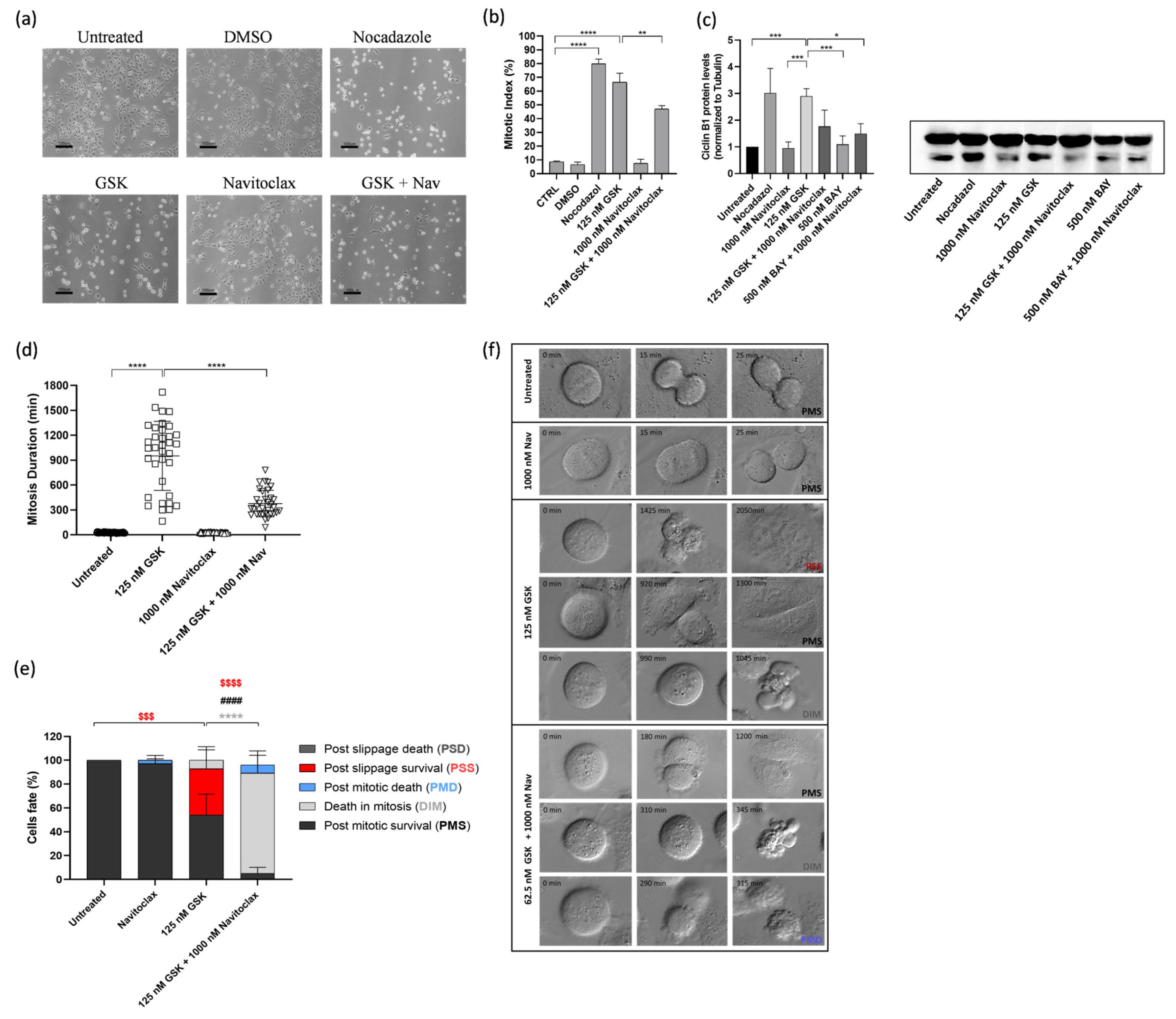
3.3. Navitoclax Prevents Mitotic Slippage Caused by GSK923295 Treatment by Accelerating Cell Death during Mitosis
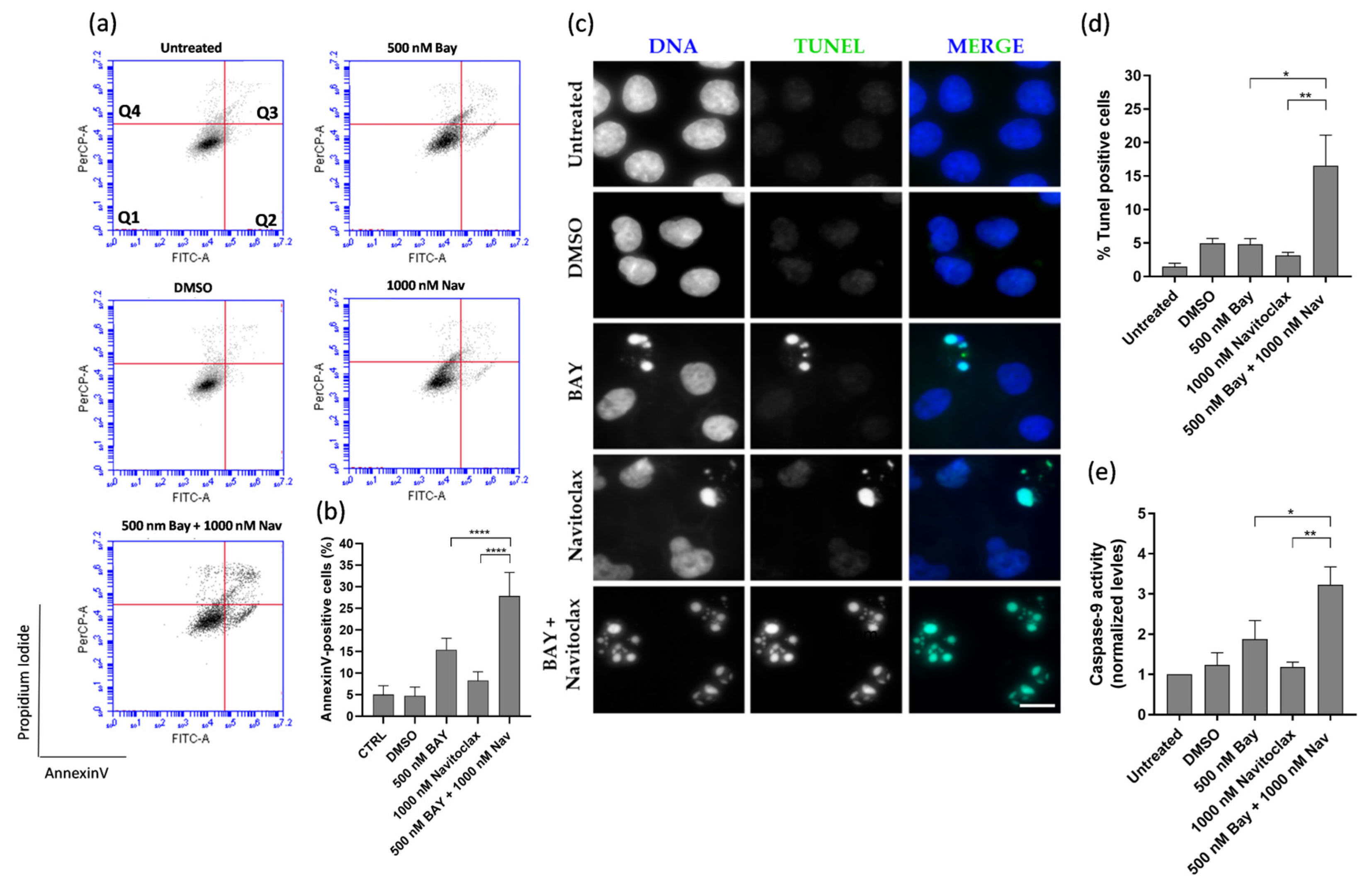
3.4. Navitoclax Prevents Post-Mitotic Survival Induced by BAY1217389 by Enhancing Post-Mitotic Death, but Only Partially
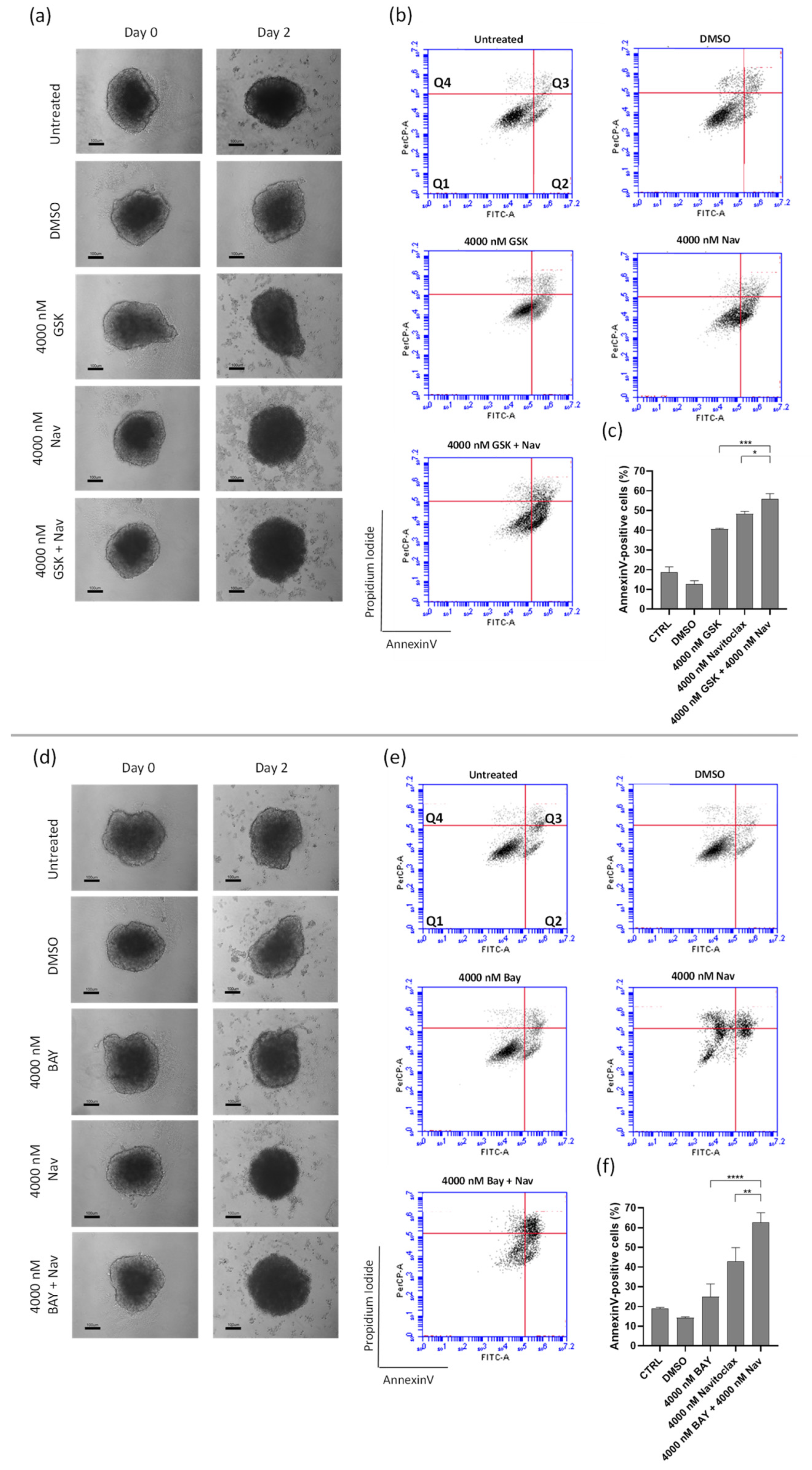
3.5. Navitoclax Sensitizes 3D Lung Cancer Spheroids to GSK923295 and BAY1217389 Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pilkington, G.; Boland, A.; Brown, T.; Oyee, J.; Bagust, A.; Dickson, R. A systematic review of the clinical effectiveness of first-line chemotherapy for adult patients with locally advanced or metastatic non-small cell lung cancer. Thorax 2015, 70, 359–367. [Google Scholar] [CrossRef]
- Institute, N.C. SEER Cancer Statistics Review (CSR) 1975–2016. Available online: https://seer.cancer.gov/archive/csr/1975_2016/ (accessed on 19 October 2023).
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Zhang, Y.; Etxeberria, J.; Arnold, M.; Cai, X.; Hao, Y.; Zou, H. Projections of Lung Cancer Incidence by 2035 in 40 Countries Worldwide: Population-Based Study. JMIR Public Health Surveill. 2023, 9, e43651. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.C.; Ribeiro, D.; Pedrosa, J.; Sarmento, B.; Silva, P.M.A.; Bousbaa, H. Mitosis inhibitors in anticancer therapy: When blocking the exit becomes a solution. Cancer Lett. 2019, 440–441, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Broggini-Tenzer, A.; Vuong, V.; Messikommer, A.; Nytko, K.J.; Guckenberger, M.; Bachmann, F.; Lane, H.A.; Pruschy, M. The novel microtubule targeting agent BAL101553 in combination with radiotherapy in treatment-refractory tumor models. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2017, 124, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.C.; Li, L.W.; Pan, S.H.; Fang, J.M.; Liu, J.H.; Cheng, T.J.; Wang, C.J.; Hung, P.F.; Chen, H.Y.; Hong, T.M.; et al. Purine-Type Compounds Induce Microtubule Fragmentation and Lung Cancer Cell Death through Interaction with Katanin. J. Med. Chem. 2016, 59, 8521–8534. [Google Scholar] [CrossRef]
- Novais, P.; Silva, P.M.A.; Amorim, I.; Bousbaa, H. Second-generation antimitotics in cancer clinical trials. Pharmaceutics 2021, 13, 1011. [Google Scholar] [CrossRef]
- Schöffski, P.; Awada, A.; de la Bigne, A.M.; Felloussi, Z.; Burbridge, M.; Cantero, F.; Colombo, R.; Maruzzelli, S.; Ammattatelli, K.; de Jonge, M.; et al. First-in-man, first-in-class phase I study with the monopolar spindle 1 kinase inhibitor S81694 administered intravenously in adult patients with advanced, metastatic solid tumours. Eur. J. Cancer 2022, 169, 135–145. [Google Scholar] [CrossRef]
- Chung, V.; Heath, E.I.; Schelman, W.R.; Johnson, B.M.; Kirby, L.C.; Lynch, K.M.; Botbyl, J.D.; Lampkin, T.A.; Holen, K.D. First-time-in-human study of GSK923295, a novel antimitotic inhibitor of centromere-associated protein E (CENP-E), in patients with refractory cancer. Cancer Chemother. Pharmacol. 2012, 69, 733–741. [Google Scholar] [CrossRef]
- Raab, M.; Krämer, A.; Hehlgans, S.; Sanhaji, M.; Kurunci-Csacsko, E.; Dötsch, C.; Bug, G.; Ottmann, O.; Becker, S.; Pachl, F.; et al. Mitotic arrest and slippage induced by pharmacological inhibition of Polo-like kinase 1. Mol. Oncol. 2015, 9, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. Cancer Cells Display Profound Intra- and Interline Variation following Prolonged Exposure to Antimitotic Drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic slippage: An old tale with a new twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef]
- Serrano-del Valle, A.; Reina-Ortiz, C.; Benedi, A.; Anel, A.; Naval, J.; Marzo, I. Future prospects for mitosis-targeted antitumor therapies. Biochem. Pharmacol. 2021, 190, 114655. [Google Scholar] [CrossRef] [PubMed]
- Partscht, P.; Simon, A.; Chen, N.P.; Erhardt, S.; Schiebel, E. The HIPK2/CDC14B-MeCP2 axis enhances the spindle assembly checkpoint block by promoting cyclin B translation. Sci. Adv. 2023, 9, eadd6982. [Google Scholar] [CrossRef] [PubMed]
- Ghelli Luserna di Rorà, A.; Martinelli, G.; Simonetti, G. The balance between mitotic death and mitotic slippage in acute leukemia: A new therapeutic window? J. Hematol. Oncol. 2019, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.M.; Ribeiro, N.; Lima, R.T.; Andrade, C.; Diogo, V.; Teixeira, J.; Florindo, C.; Tavares, Á.; Vasconcelos, M.H.; Bousbaa, H. Suppression of spindly delays mitotic exit and exacerbates cell death response of cancer cells treated with low doses of paclitaxel. Cancer Lett. 2017, 394, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Klener, P.; Sovilj, D.; Renesova, N.; Andera, L. BH3 Mimetics in Hematologic Malignancies. Int. J. Mol. Sci. 2021, 22, 10157. [Google Scholar] [CrossRef]
- Opydo-Chanek, M.; Gonzalo, O.; Marzo, I. Multifaceted anticancer activity of BH3 mimetics: Current evidence and future prospects. Biochem. Pharmacol. 2017, 136, 12–23. [Google Scholar] [CrossRef]
- Pinto, B.; Novais, P.; Henriques, A.C.; Carvalho-Tavares, J.; Silva, P.M.A.; Bousbaa, H. Navitoclax Enhances the Therapeutic Effects of PLK1 Targeting on Lung Cancer Cells in 2D and 3D Culture Systems. Pharmaceutics 2022, 14, 1209. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.; Sloss, O.; Topham, C.; Nelson, L.; Tighe, A.; Taylor, S.S. Inhibition of Bcl-xL sensitizes cells to mitotic blockers, but not mitotic drivers. Open Biol. 2016, 6, 160134. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.C.; Silva, P.M.A.; Sarmento, B.; Bousbaa, H. Antagonizing the spindle assembly checkpoint silencing enhances paclitaxel and Navitoclax-mediated apoptosis with distinct mechanistic. Sci. Rep. 2021, 11, 4139. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.W.; Lad, L.; Luo, L.; Qian, X.; Knight, S.D.; Nevins, N.; Brejc, K.; Sutton, D.; Gilmartin, A.G.; Chua, P.R.; et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc. Natl. Acad. Sci. USA 2010, 107, 5839–5844. [Google Scholar] [CrossRef] [PubMed]
- Wengner, A.M.; Siemeister, G.; Koppitz, M.; Schulze, V.; Kosemund, D.; Klar, U.; Stoeckigt, D.; Neuhaus, R.; Lienau, P.; Bader, B.; et al. Novel Mps1 kinase inhibitors with potent antitumor activity. Mol. Cancer Ther. 2016, 15, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Heuser, J.E.; Waterman, C.M.; Cleveland, D.W. CENP-E combines a slow, processive motor and a flexible coiled coil to produce an essential motile kinetochore tether. J. Cell Biol. 2008, 181, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.; Bechi, B.; Tighe, A.; Thompson, S.; Procter, D.J.; Taylor, S.S. Cenp-E inhibitor GSK923295: Novel synthetic route and use as a tool to generate aneuploidy. Oncotarget 2015, 6, 20921–20932. [Google Scholar] [CrossRef]
- Pachis, S.T.; Kops, G. Leader of the SAC: Molecular mechanisms of Mps1/TTK regulation in mitosis. Open Biol. 2018, 8, 180109. [Google Scholar] [CrossRef]
- Tipton, A.R.; Ji, W.; Sturt-Gillespie, B.; Bekier, M.E., 2nd; Wang, K.; Taylor, W.R.; Liu, S.T. Monopolar spindle 1 (MPS1) kinase promotes production of closed MAD2 (C-MAD2) conformer and assembly of the mitotic checkpoint complex. J. Biol. Chem. 2013, 288, 35149–35158. [Google Scholar] [CrossRef]
- Mason, J.M.; Wei, X.; Fletcher, G.C.; Kiarash, R.; Brokx, R.; Hodgson, R.; Beletskaya, I.; Bray, M.R.; Mak, T.W. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3127–3132. [Google Scholar] [CrossRef]
- Ling, Y.; Zhang, X.; Bai, Y.; Li, P.; Wei, C.; Song, T.; Zheng, Z.; Guan, K.; Zhang, Y.; Zhang, B.; et al. Overexpression of Mps1 in colon cancer cells attenuates the spindle assembly checkpoint and increases aneuploidy. Biochem. Biophys. Res. Commun. 2014, 450, 1690–1695. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Qu, T. Expression of CENPE and its prognostic role in non-small cell lung cancer. Open Med. 2019, 14, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Chen, Z.; Kawakami, M.; Chen, Y.; Roszik, J.; Mustachio, L.M.; Kurie, J.M.; Villalobos, P.; Lu, W.; Behrens, C.; et al. Tyrosine threonine kinase inhibition eliminates lung cancers by augmenting apoptosis and polyploidy. Mol. Cancer Ther. 2019, 18, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Yuan, F.; Zhao, Z.; Xue, T.; Ge, N.; Ren, Z.; Zhang, L. Expression and Clinical Significance of MPS-1 in Hepatocellular Carcinoma. Int. J. Gen. Med. 2021, 14, 9145–9152. [Google Scholar] [CrossRef]
- Landi, M.T.; Dracheva, T.; Rotunno, M.; Figueroa, J.D.; Liu, H.; Dasgupta, A.; Mann, F.E.; Fukuoka, J.; Hames, M.; Bergen, A.W.; et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE 2008, 3, e1651. [Google Scholar] [CrossRef]
- Naranjo, S.; Cabana, C.M.; LaFave, L.M.; Romero, R.; Shanahan, S.L.; Bhutkar, A.; Westcott, P.M.K.; Schenkel, J.M.; Ghosh, A.; Liao, L.Z.; et al. Modeling diverse genetic subtypes of lung adenocarcinoma with a next-generation alveolar type 2 organoid platform. Genes Dev. 2022, 36, 936–949. [Google Scholar] [CrossRef]
- Mohamad Anuar, N.N.; Nor Hisam, N.S.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 564108. [Google Scholar] [CrossRef]
- Nakayama, Y.; Yamaguchi, N. Role of cyclin B1 levels in DNA damage and DNA damage-induced senescence. Int. Rev. Cell Mol. Biol. 2013, 305, 303–337. [Google Scholar] [CrossRef]
- Würstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res. 2012, 318, 1213–1220. [Google Scholar] [CrossRef]
- Song, Z.; Chen, Y.; Chang, H.; Guo, Y.; Gao, Q.; Wei, Z.; Gong, L.; Zhang, G.; Zheng, Z. Rhein suppresses African swine fever virus replication in vitro via activating the caspase-dependent mitochondrial apoptosis pathway. Virus Res. 2023, 338, 199238. [Google Scholar] [CrossRef]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.A.; Marcato, P.D.; Mondal, A.; Chowdhury, N.; Gebeyehu, A.; Surapaneni, S.K.; Bentley, M.; Amaral, R.; Pan, C.X.; Singh, M. Cytotoxic and chemosensitizing effects of glycoalkaloidic extract on 2D and 3D models using RT4 and patient derived xenografts bladder cancer cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 119, 111460. [Google Scholar] [CrossRef] [PubMed]
- Samimi, H.; Sohi, A.N.; Irani, S.; Arefian, E.; Mahdiannasser, M.; Fallah, P.; Haghpanah, V. Alginate-based 3D cell culture technique to evaluate the half-maximal inhibitory concentration: An in vitro model of anticancer drug study for anaplastic thyroid carcinoma. Thyroid Res. 2021, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Kerslake, R.; Belay, B.; Panfilov, S.; Hall, M.; Kyrou, I.; Randeva, H.S.; Hyttinen, J.; Karteris, E.; Sisu, C. Transcriptional Landscape of 3D vs. 2D Ovarian Cancer Cell Models. Cancers 2023, 15, 3350. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | IC50 (nM) |
|---|---|
| GSK923295 | 150 ± 30 |
| Navitoclax | 13,050 ± 690 |
| Drugs | IC50 (nM) |
|---|---|
| BAY1217389 | 4340 ± 60 |
| Navitoclax | 13,310 ± 910 |
| Drugs | IC50 (nM) |
|---|---|
| GSK923295 | >16,000 |
| navitoclax | 6480 ± 1070 |
| Drugs | IC50 (nM) |
|---|---|
| BAY1217389 | >16,000 |
| navitoclax | 6340 ± 930 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, B.; Silva, J.P.N.; Silva, P.M.A.; Barbosa, D.J.; Sarmento, B.; Tavares, J.C.; Bousbaa, H. Maximizing Anticancer Response with MPS1 and CENPE Inhibition Alongside Apoptosis Induction. Pharmaceutics 2024, 16, 56. https://doi.org/10.3390/pharmaceutics16010056
Pinto B, Silva JPN, Silva PMA, Barbosa DJ, Sarmento B, Tavares JC, Bousbaa H. Maximizing Anticancer Response with MPS1 and CENPE Inhibition Alongside Apoptosis Induction. Pharmaceutics. 2024; 16(1):56. https://doi.org/10.3390/pharmaceutics16010056
Chicago/Turabian StylePinto, Bárbara, João P. N. Silva, Patrícia M. A. Silva, Daniel José Barbosa, Bruno Sarmento, Juliana Carvalho Tavares, and Hassan Bousbaa. 2024. "Maximizing Anticancer Response with MPS1 and CENPE Inhibition Alongside Apoptosis Induction" Pharmaceutics 16, no. 1: 56. https://doi.org/10.3390/pharmaceutics16010056
APA StylePinto, B., Silva, J. P. N., Silva, P. M. A., Barbosa, D. J., Sarmento, B., Tavares, J. C., & Bousbaa, H. (2024). Maximizing Anticancer Response with MPS1 and CENPE Inhibition Alongside Apoptosis Induction. Pharmaceutics, 16(1), 56. https://doi.org/10.3390/pharmaceutics16010056