

l-Asparaginase Type II from Fusarium proliferatum: Heterologous Expression and In Silico Analysis

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Fungal, Bacterial Strains, and Plasmids
2.3. Microorganisms and Growth Conditions
2.4. Sequencing of F. proliferatum l-Asparaginase Gene
2.5. In Silico Analysis
2.6. Cloning of l-Asparaginase Gene from F. proliferatum
2.7. Heterologous l-Asparaginase Expression
2.8. Cell Disruption Tests
2.9. l-Asparaginase Activity
2.10. l-Asparaginase Purification
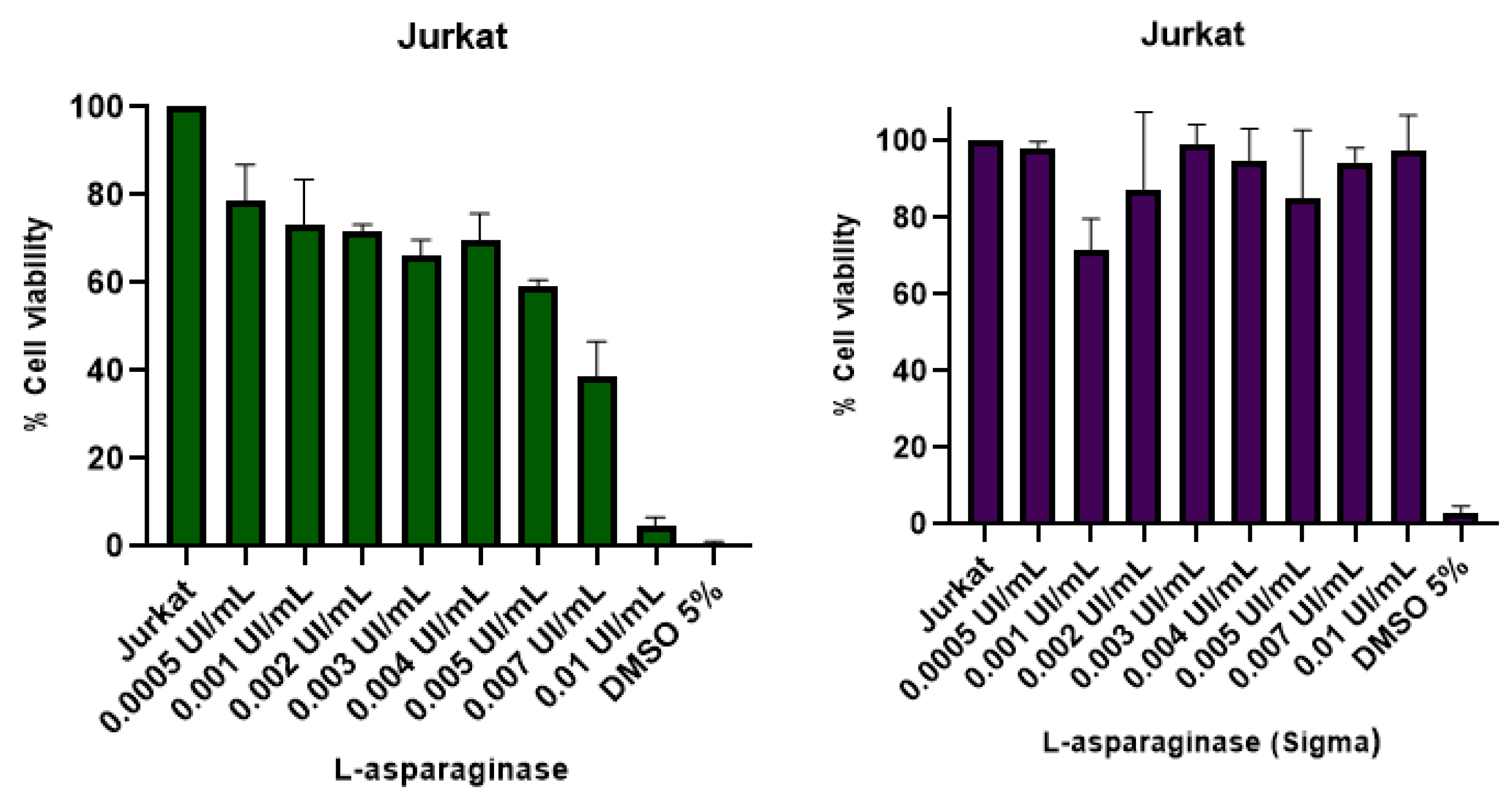
2.11. Cytotoxic Effect Using MTT Assay
3. Results
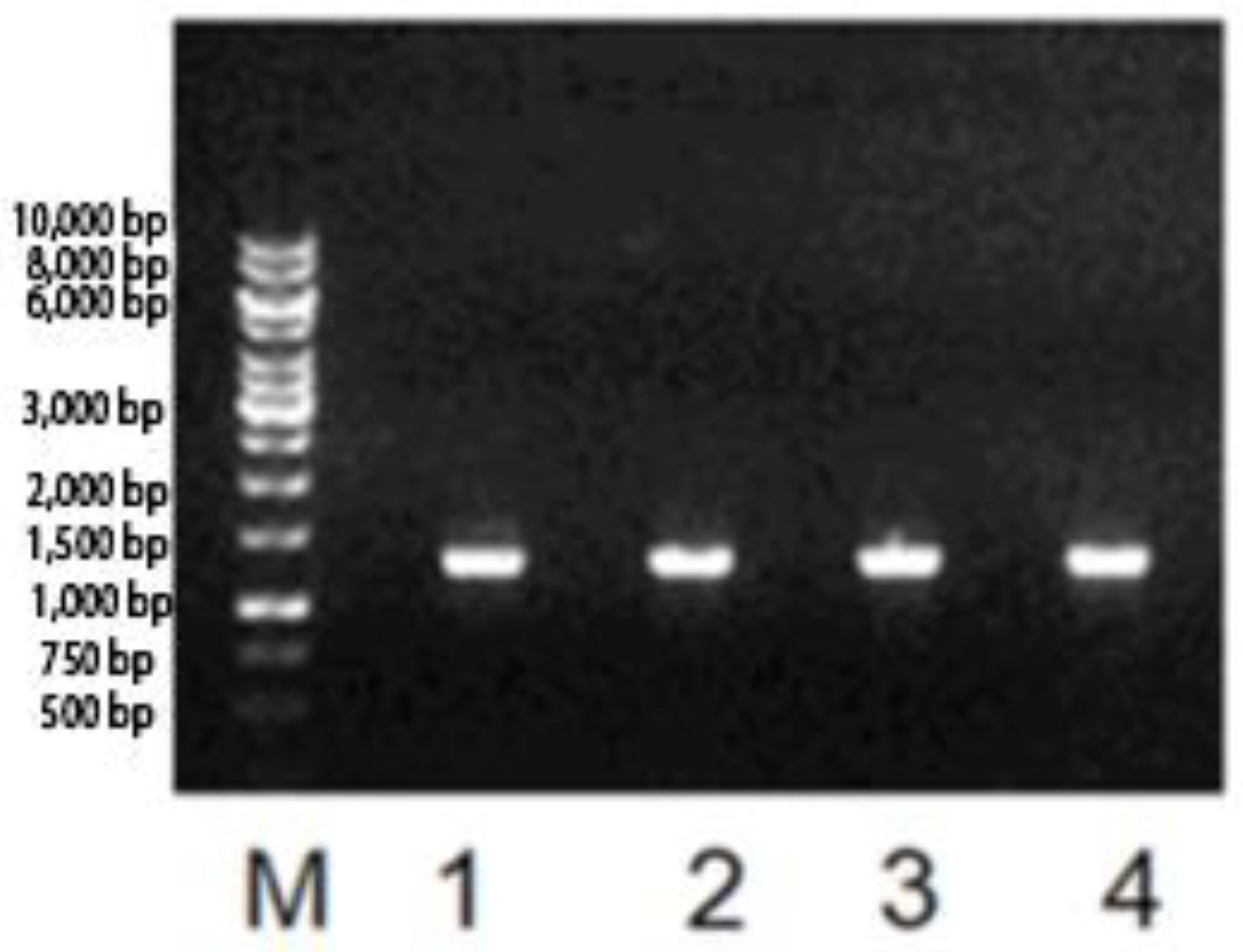
3.1. l-Asparaginase Gene Identification
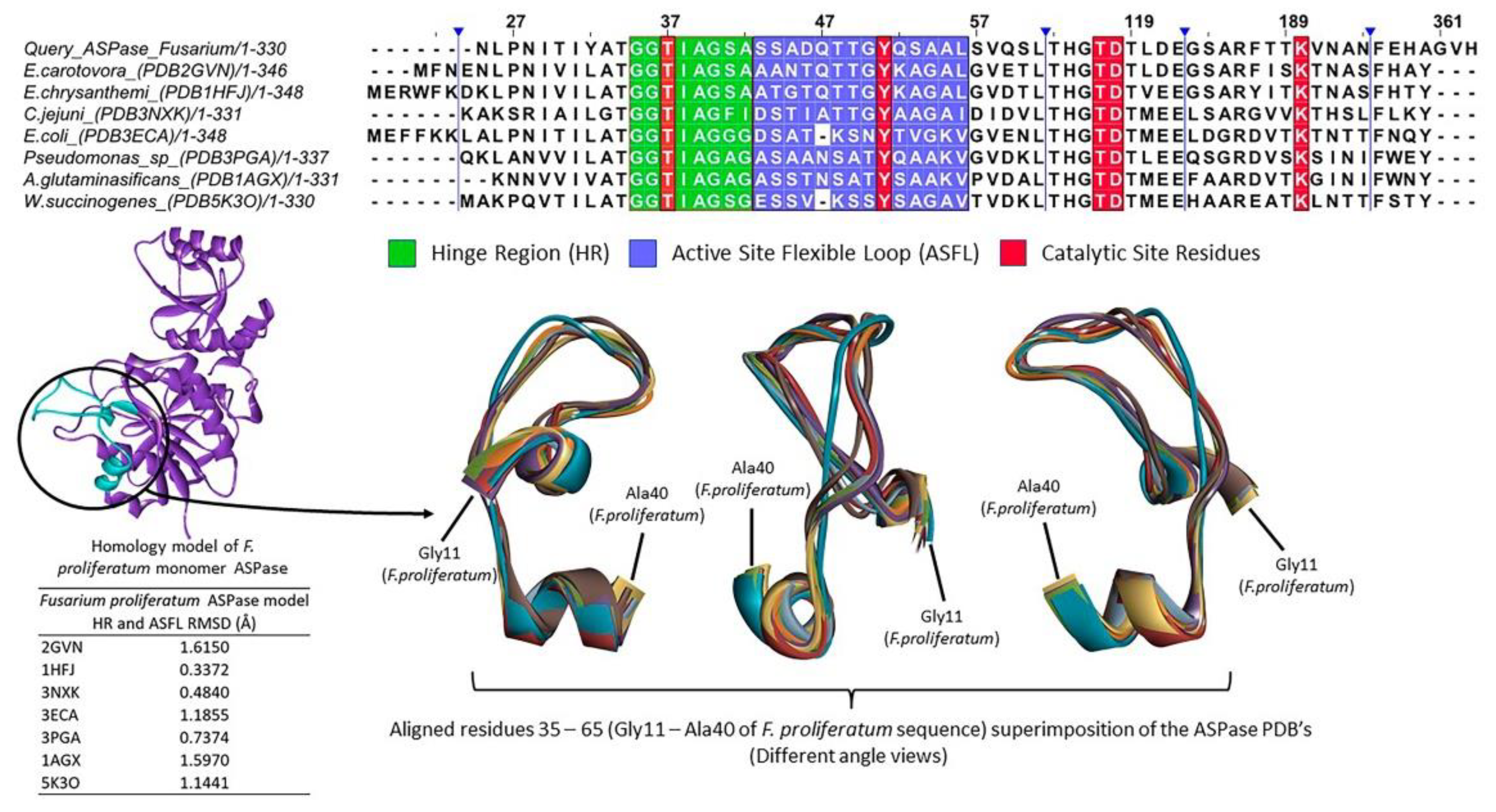
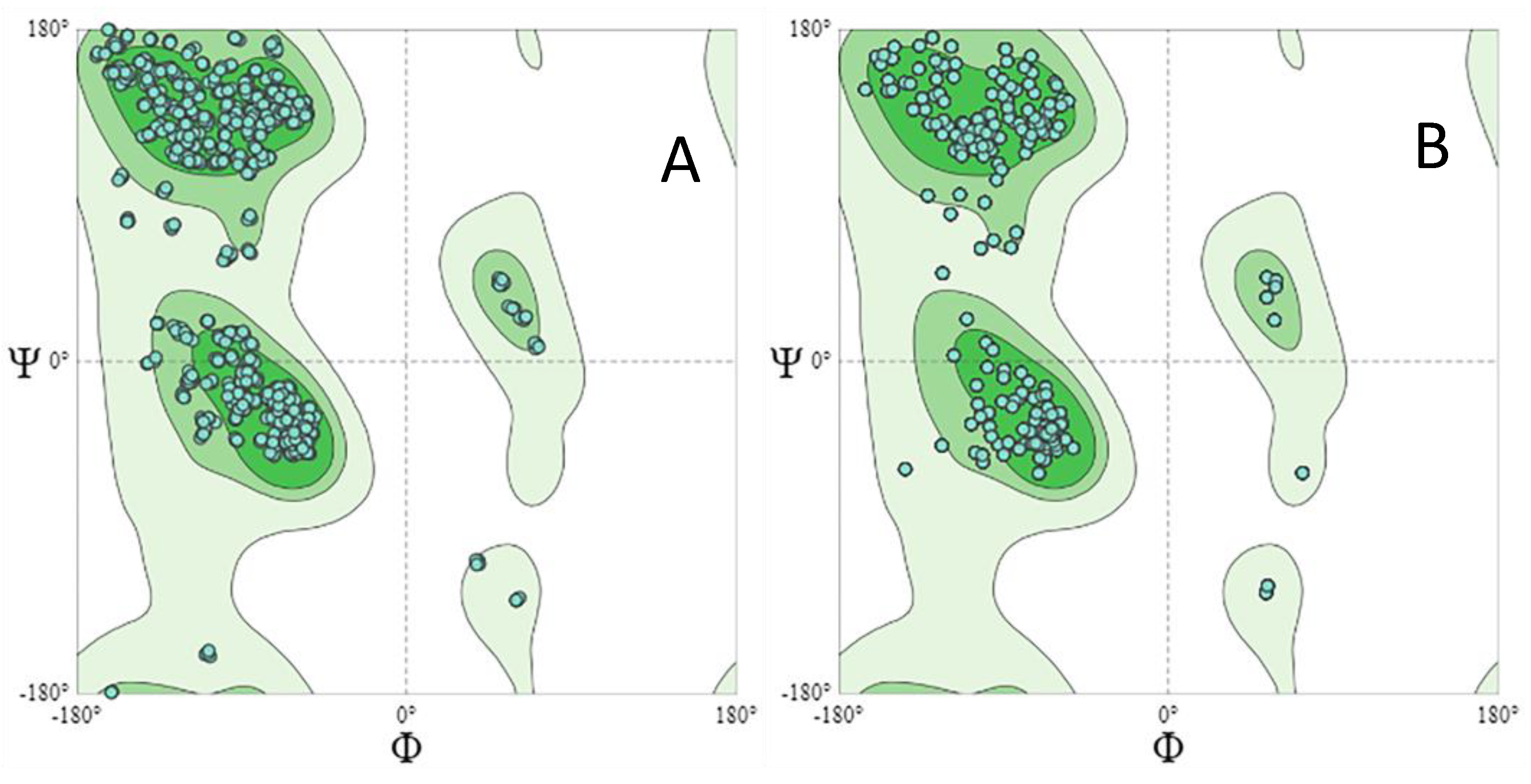
3.2. In Silico Analysis
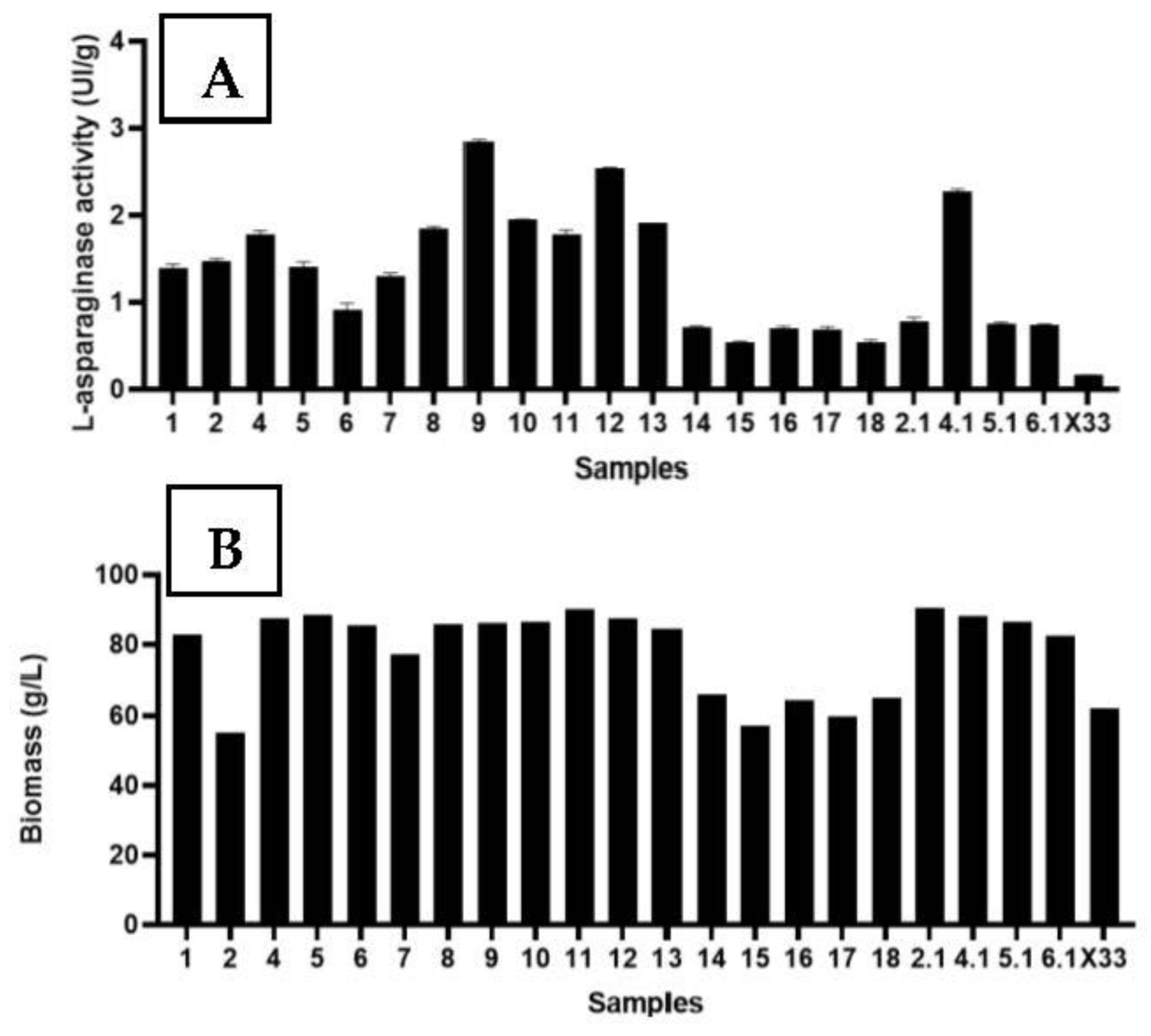
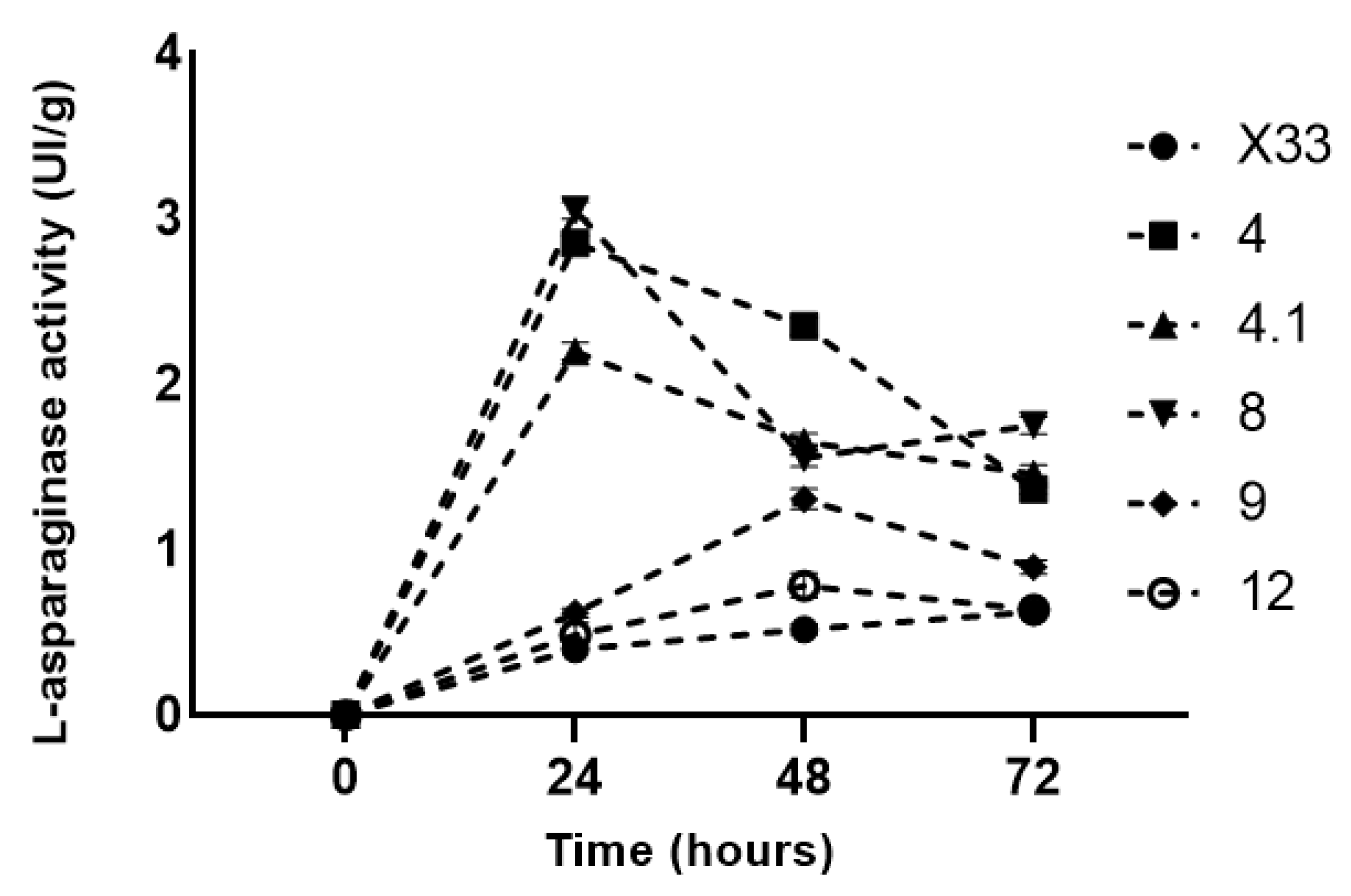
3.3. Heterologous l-Asparaginase Activity
3.4. Growth Curve
3.5. Cell Disruption Test
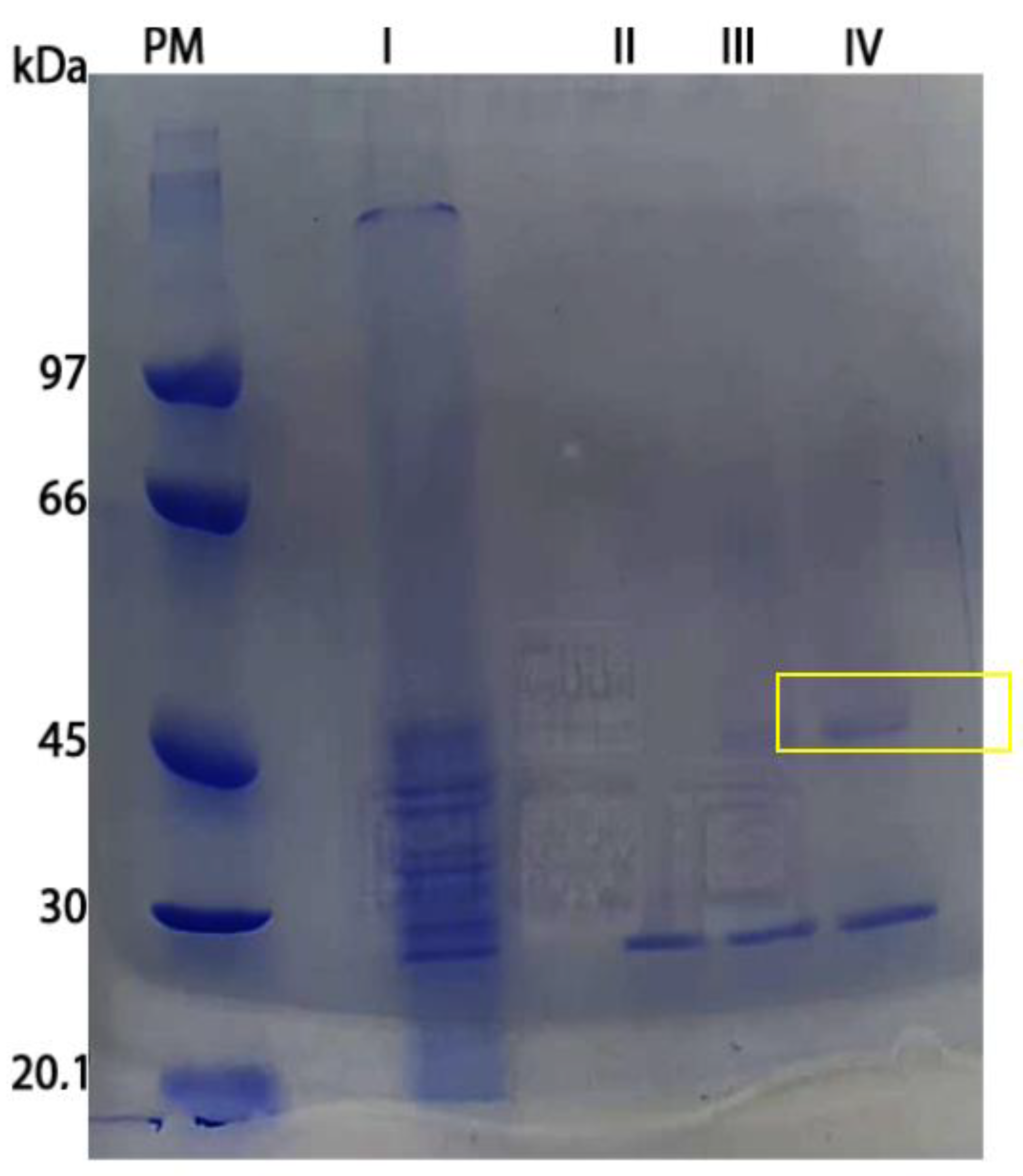
3.6. l-Asparaginase Partial Purification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darvishi, F.; Jahanafrooz, Z.; Mokhtarzadeh, A. Microbial L-asparaginase as a promising enzyme for treatment of various cancers. Appl. Microbiol. Biotechnol. 2022, 106, 5335–5347. [Google Scholar] [CrossRef] [PubMed]
- Dhankhar, R.; Gupta, V.; Kumar, S.; Kapoor, R.K.; Gulati, P. Microbial enzymes for deprivation of amino acid metabolism in malignant cells: Biological strategy for cancer treatment. Appl. Microbiol. Biotechnol. 2020, 104, 2857–2869. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.R.; Byregowda, S.M.; Veeregowda, B.M.; Balamurugan, V. An overview of heterologous expression host systems for the production of recombinant proteins. Adv. Anim. Vet. Sci. 2016, 4, 346–356. [Google Scholar] [CrossRef]
- Freitas, M.; Souza, P.; Homem-De-Mello, M.; Fonseca-Bazzo, Y.M.; Silveira, D.; Filho, E.X.F.; Junior, A.P.; Sarker, D.; Timson, D.; Inácio, J.; et al. L-Asparaginase from Penicillium sizovae Produced by a Recombinant Komagataella phaffii Strain. Pharmaceuticals 2022, 15, 746. [Google Scholar] [CrossRef]
- Looser, V.; Bruhlmann, B.; Bumbak, F.; Stenger, C.; Costa, M.; Camattari, A.; Fotiadis, D.; Kovar, K. Cultivation strategies to enhance productivity of Pichia pastoris: A review. Biotechnol. Adv. 2015, 33, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Vieira Gomes, A.M.; Souza Carmo, T.; Silva Carvalho, L.; Mendonça Bahia, F.; Parachin, N.S. Comparison of yeasts as hosts for recombinant protein production. Microorganisms 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Vieira, S.M.; da Rocha, S.L.G.; Neves-Ferreira, A.G.d.C.; Almeida, R.V.; Perales, J. Heterologous expression of the antimyotoxic protein DM64 in Pichia pastoris. PLoS Neglected Trop. Dis. 2017, 11, e0005829. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Alper, H.S. Synthetic biology and molecular genetics in non-conventional yeasts: Current tools and future advances. Fungal Genet. Biol. 2016, 89, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Cereghino, J.L.; Cregg, J.M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 2000, 24, 45–66. [Google Scholar] [CrossRef]
- Tschopp, J.F.; Brust, P.F.; Cregg, J.M.; Stillman, C.A.; Gingeras, T.R. Expression of the lacZ gene from two methanol-regulated promoters in Pichia pastoris. Nucleic Acids Res. 1987, 15, 3859–3876. [Google Scholar] [CrossRef]
- Katsonis, P.; Wilhelm, K.; Williams, A.; Lichtarge, O. Genome interpretation using in silico predictors of variant impact. Hum. Genet. 2022, 141, 1549–1577. [Google Scholar] [CrossRef] [PubMed]
- Ganorkar, S.B.; Vander Heyden, Y. Recent trends in pharmaceutical analysis to foster modern drug discovery by comparative in-silico profiling of drugs and related substances. TrAC Trends Anal. Chem. 2022, 157, 116747. [Google Scholar] [CrossRef]
- Freitas, M.; Souza, P.; Cardoso, S.; Cruvinel, K.; Abrunhosa, L.S.; Ferreira Filho, E.X.; Inácio, J.; Pinho, D.B.; Pessoa, A.; Magalhães, P.O. Filamentous Fungi Producing l-Asparaginase with Low Glutaminase Activity Isolated from Brazilian Savanna Soil. Pharmaceutics 2021, 13, 1268. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Tuziuti, T.; Yasui, K.; Kozuka, T.; Towata, A. Protein release from yeast cells as an evaluation method of physical effects in ultrasonic field. Ultrason. Sonochem. 2008, 15, 995–1000. [Google Scholar] [CrossRef]
- Drainas, C.; Kinghorn, J.R.; Pateman, J.A. Aspartic hydroxamate resistance and asparaginase regulation in the fungus Aspergillus nidulans. J. Gen. Microbiol. 1977, 98, 493–501. [Google Scholar] [CrossRef]
- Laemmli, U. Cleavage of structural proteins during the assembly of head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- El-Gendy, M.M.A.A.; Awad, M.F.; El-Shenawy, F.S.; El-Bondkly, A.M.A. Production, purification, characterization, antioxidant and antiproliferative activities of extracellular L-asparaginase produced by Fusarium equiseti AHMF4. Saudi J. Biol. Sci. 2021, 28, 2540–2548. [Google Scholar] [CrossRef]
- Jebur, I.M.; Najaf, K.N.; Ali HH, R.; Ibrahim, M.S. Cloning and expression of l-Asparaginase gene from Aspergillus terreus in E. coli. J. Pharm. Sci. Res. 2019, 11, 961–965. [Google Scholar]
- Saeed, H.; Ali, H.; Soudan, H.; Embaby, A.; El-Sharkawy, A.; Farag, A.; Hussein, A.; Ataya, F. Molecular cloning, structural modeling and production of recombinant Aspergillus terreus L. asparaginase in Escherichia coli. Int. J. Biol. Macromol. 2018, 106, 1041–1051. [Google Scholar] [CrossRef]
- Kot, A.M.; Gientka, I.; Bzducha-Wróbel, A.; Błażejak, S.; Kurcz, A. Comparison of simple and rapid cell wall disruption methods for improving lipid extraction from yeast cells. J. Microbiol. Methods 2020, 176, 105999. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.A.; Severino, N.M.B.; Mansure, J.J.; Martins, A.S.; Oliveira, E.M.M.; Siani, A.C.; Pereira, N., Jr.; Torres, F.A.G.; Bon, E.P.S. Asparaginase production by a recombinant Pichia pastoris strain harbouring Saccharomyces cerevisiae ASP3 gene. Enzym. Microb. Technol. 2006, 39, 1457–1463. [Google Scholar] [CrossRef]
- Dwivedi, V.D.; Mishra, S.K. In silico analysis of L-asparaginase from different source organisms. Interdiscip. Sci. Comput. Life Sci. 2014, 6, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Lima, G.M.; Effer, B.; Biasoto, H.P.; Feijoli, V.; Pessoa, A.; Palmisano, G.; Monteiro, G. Glycosylation of L-asparaginase from E. coli through yeast expression and site-directed mutagenesis. Biochem. Eng. J. 2020, 156, 107516. [Google Scholar] [CrossRef]
- Sajitha, S.; Vidya, J.; Varsha, K.; Binod, P.; Pandey, A. Cloning and expression of l-asparaginase from E. coli in eukaryotic expression system. Biochem. Eng. J. 2015, 102, 14–17. [Google Scholar] [CrossRef]
- Arumugam, N.; Thangavelu, P. Purification and anticancer activity of glutaminase and urease free intracellular l-asparaginase from Chaetomium sp. Protein Expr. Purif. 2022, 190, 106006. [Google Scholar] [CrossRef]
- Jozala, A.F.; Geraldes, D.C.; Tundisi, L.L.; Feitosa, V.d.A.; Breyer, C.A.; Cardoso, S.L.; Mazzola, P.G.; de Oliveira-Nascimento, L.; Rangel-Yagui, C.d.O.; Magalhães, P.d.O.; et al. Biopharmaceuticals from microorganisms: From production to purification. Braz. J. Microbiol. 2016, 47, 51–63. [Google Scholar] [CrossRef]
- Kleingesinds, E.K.; Parizotto, L.d.A.; Effer, B.; Monteiro, G.; Long, P.F.; Arroyo-Berdugo, Y.; Behrends, V.; Esposito, M.T.; Calle, Y.; Pessoa, A., Jr. Downstream process and evaluation of the concomitant impact of a recombinant glycosylated L-asparaginase on leukemic cancer cells and the bone marrow tumor microenvironment. Process. Biochem. 2023, 131, 41–51. [Google Scholar] [CrossRef]
- Yap, L.S.; Lee, W.L.; Ting, A.S.Y. Optimization of L-asparaginase production from endophytic Fusarium proliferatum using OFAT and RSM and its cytotoxic evaluation. J. Microbiol. Methods 2021, 191, 106358. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purification Step | Volume (mL) | Total Activity (U/mL) | Total Protein (mg) | Specific Activity (U/mg) | Purification Fold | Yield (%) |
|---|---|---|---|---|---|---|
| Crude Extract | 25 | 2.3 | 2.2 | 1.04 | 1.00 | 100 |
| DEAE FF | 25 | 0.09 | 0.29 | 0.31 | 0.29 | 7.25 |
| Concentrated Enzyme (10×) | 2.5 | 1.16 | 0.94 | 1.23 | 1.18 | 2.95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, S.L.; Souza, P.M.; Rodrigues, K.; Mota, I.d.S.; Filho, E.F.; Fávaro, L.C.d.L.; Saldanha-Araujo, F.; Homem-de-Mello, M.; Pessoa, A.; Silveira, D.; et al. l-Asparaginase Type II from Fusarium proliferatum: Heterologous Expression and In Silico Analysis. Pharmaceutics 2023, 15, 2352. https://doi.org/10.3390/pharmaceutics15092352
Cardoso SL, Souza PM, Rodrigues K, Mota IdS, Filho EF, Fávaro LCdL, Saldanha-Araujo F, Homem-de-Mello M, Pessoa A, Silveira D, et al. l-Asparaginase Type II from Fusarium proliferatum: Heterologous Expression and In Silico Analysis. Pharmaceutics. 2023; 15(9):2352. https://doi.org/10.3390/pharmaceutics15092352
Chicago/Turabian StyleCardoso, Samuel Leite, Paula Monteiro Souza, Kelly Rodrigues, Isabella de Souza Mota, Edivaldo Ferreira Filho, Léia Cecilia de Lima Fávaro, Felipe Saldanha-Araujo, Mauricio Homem-de-Mello, Adalberto Pessoa, Dâmaris Silveira, and et al. 2023. "l-Asparaginase Type II from Fusarium proliferatum: Heterologous Expression and In Silico Analysis" Pharmaceutics 15, no. 9: 2352. https://doi.org/10.3390/pharmaceutics15092352
APA StyleCardoso, S. L., Souza, P. M., Rodrigues, K., Mota, I. d. S., Filho, E. F., Fávaro, L. C. d. L., Saldanha-Araujo, F., Homem-de-Mello, M., Pessoa, A., Silveira, D., Fonseca-Bazzo, Y. M., & Magalhães, P. O. (2023). l-Asparaginase Type II from Fusarium proliferatum: Heterologous Expression and In Silico Analysis. Pharmaceutics, 15(9), 2352. https://doi.org/10.3390/pharmaceutics15092352