
Effect of Gut Microbiota on the Pharmacokinetics of Nifedipine in Spontaneously Hypertensive Rats


Abstract
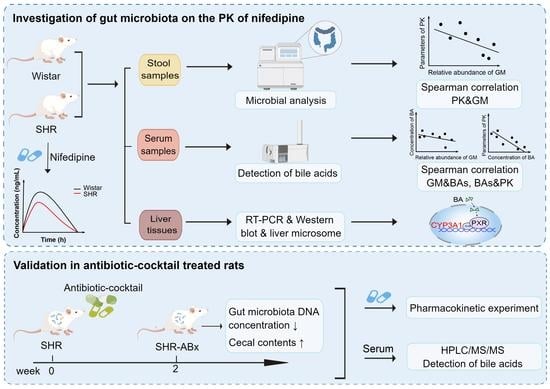
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Pharmacokinetic Study in Wistar Rats and SHRs
2.2.1. Animals
2.2.2. Pharmacokinetic Experiments
2.2.3. UPLC-MS/MS
2.3. Gut Microbiome Analysis
2.4. Analysis of Bile Acids
2.5. RNA Extraction and Quantitative Real-Time PCR
2.6. Western Blot
2.7. Microsomal Preparation and Enzyme Activity Detection
2.8. Primary Hepatocyte Isolation and Induction
2.9. Pharmacokinetic Study in Antibiotic-Cocktail-Treated Rats
2.10. Blood Biochemistry Test and Histological Assays
2.11. Statistical Analysis
3. Results
3.1. Oral Bioavailability of Nifedipine Decreased in SHRs
3.2. Gut Microbial Dysbiosis and Bacteroides Enrichment in SHRs
3.3. Association between Gut Microbiota and Pharmacokinetic Parameters
3.4. Perturbations of the Microbiota Altered the BA Profiles in SHRs
3.5. Gut Microbiota Altered the Expression and Enzyme Activity of CYP3A1
3.6. Correlation among Microbiota, Bile Acids, and Pharmacokinetic Parameters
3.7. Using the Antibiotic-Cocktail-Treated Rats to Confirm That Gut Microbiota Are Involved in the Metabolism of Nifedipine
3.8. Effect of Liver and Renal Functions on Drug Metabolism and Intestinal Functions on Drug Absorption
4. Discussion
5. Limitations of This Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sorkin, E.M.; Clissold, S.P.; Brogden, R.N. Nifedipine A Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy, in Ischaemic Heart Disease, Hypertension and Related Cardiovascular Disorders. Drug Eval. 1985, 30, 182–274. [Google Scholar] [CrossRef]
- Nader, A.M.; Quinney, S.K.; Fadda, H.M.; Foster, D.R. Effect of Gastric Fluid Volume on the In Vitro Dissolution and In Vivo Absorption of BCS Class II Drugs: A Case Study with Nifedipine. AAPS J. 2016, 18, 981–988. [Google Scholar] [CrossRef]
- Wang, H.J.; Lu, C.K.; Chen, W.C.; Chen, A.C.; Ueng, Y.F. Shenmai-Yin decreased the clearance of nifedipine in rats: The involvement of time-dependent inhibition of nifedipine oxidation. J. Food Drug Anal. 2019, 27, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Krecic-Shepard, M.E.; Park, K.; Barnas, C.; Slimko, J.; Kerwin, D.R.; Schwartz, J.B. Race and sex influence clearance of nifedipine: Results of a population study. Clin. Pharmacol. Ther. 2000, 68, 130–142. [Google Scholar] [CrossRef]
- Littler, W.A. Control of blood pressure in hypertensive patients with felodipine extended release or nifedipine retard. Br. J. Clin. Pharmacol. 1990, 30, 871–878. [Google Scholar] [CrossRef]
- Guo, J.; Xu, Y.; Chen, L.-J.; Zhang, S.-X.; Liou, Y.-L.; Chen, X.-P.; Tan, Z.-R.; Zhou, H.-H.; Zhang, W.; Chen, Y. Gut microbiota and host Cyp450s co-contribute to pharmacokinetic variability in mice with non-alcoholic steatohepatitis: Effects vary from drug to drug. J. Adv. Res. 2021, 39, 319–332. [Google Scholar] [CrossRef]
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond gut feelings: How the gut microbiota regulates blood pressure. Nat. Rev. Cardiol. 2018, 15, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Javdan, B.; Lopez, J.G.; Chankhamjon, P.; Lee, Y.J.; Hull, R.; Wu, Q.; Wang, X.; Chatterjee, S.; Donia, M.S. Personalized Mapping of Drug Metabolism by the Human Gut Microbiome. Cell 2020, 181, 1661–1679.e1622. [Google Scholar] [CrossRef]
- Haiser, H.J.; Gootenberg, D.B.; Chatman, K.; Sirasani, G.; Balskus, E.P.; Turnbaugh, P.J. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 2013, 341, 295–298. [Google Scholar] [CrossRef]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 2019, 570, 462–467. [Google Scholar] [CrossRef]
- Meinl, W.; Sczesny, S.; Brigelius-Flohé, R.; Blaut, M.; Glatt, H. Impact of gut microbiota on intestinal and hepatic levels of phase 2 xenobiotic-metabolizing enzymes in the rat. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 1179–1186. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Y.; Zhou, R.; Wu, T.; Zhu, P.; Liu, Y.; Zhou, J.; Xiong, Y.; Xiong, Y.; Zhou, H.; et al. Antibiotics-induced depletion of rat microbiota induces changes in the expression of host drug-processing genes and pharmacokinetic behaviors of CYPs probe drugs. Drug Metab. Dispos. Biol. Fate Chem. 2023, 51, 509–520. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, R.; Guo, P.; Li, P.; Huang, X.; Wei, Y.; Yang, C.; Zhou, J.; Yang, T.; Liu, Y.; et al. Effects of intestinal microbiota on pharmacokinetics of cyclosporine a in rats. Front. Microbiol. 2022, 13, 1032290. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Baillie, R.A.; Zhu, H.; Zeng, Z.B.; Wiest, M.M.; Nguyen, U.T.; Wojnoonski, K.; Watkins, S.M.; Trupp, M.; Krauss, R.M. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS ONE 2011, 6, e25482. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Liddle, C.; Downes, M. FXR and PXR: Potential therapeutic targets in cholestasis. J. Steroid Biochem. Mol. Biol. 2012, 130, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, P.; Zhou, T.; Lu, J.; Spatz, E.S.; Nasir, K.; Jiang, L.; Krumholz, H.M. Comparison of Prevalence, Awareness, Treatment, and Control of Cardiovascular Risk Factors in China and the United States. J. Am. Heart Assoc. 2018, 7, e007462. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lu, Y.; Wang, X.; Li, X.; Linderman, G.C.; Wu, C.; Cheng, X.; Mu, L.; Zhang, H.; Liu, J.; et al. Prevalence, awareness, treatment, and control of hypertension in China: Data from 1·7 million adults in a population-based screening study (China PEACE Million Persons Project). Lancet 2017, 390, 2549–2558. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef]
- Yan, Q.; Gu, Y.; Li, X.; Yang, W.; Jia, L.; Chen, C.; Han, X.; Huang, Y.; Zhao, L.; Li, P.; et al. Alterations of the Gut Microbiome in Hypertension. Front. Cell Infect. Microbiol. 2017, 7, 381. [Google Scholar] [CrossRef]
- Huart, J.; Leenders, J.; Taminiau, B.; Descy, J.; Saint-Remy, A.; Daube, G.; Krzesinski, J.M.; Melin, P.; de Tullio, P.; Jouret, F. Gut Microbiota and Fecal Levels of Short-Chain Fatty Acids Differ Upon 24-Hour Blood Pressure Levels in Men. Hypertension 2019, 74, 1005–1013. [Google Scholar] [CrossRef]
- Sun, S.; Lulla, A.; Sioda, M.; Winglee, K.; Wu, M.C.; Jacobs, D.R., Jr.; Shikany, J.M.; Lloyd-Jones, D.M.; Launer, L.J.; Fodor, A.A.; et al. Gut Microbiota Composition and Blood Pressure. Hypertension 2019, 73, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef]
- Adnan, S.; Nelson, J.W.; Ajami, N.J.; Venna, V.R.; Petrosino, J.F.; Bryan, R.M., Jr.; Durgan, D.J. Alterations in the gut microbiota can elicit hypertension in rats. Physiol. Genom. 2017, 49, 96–104. [Google Scholar] [CrossRef]
- Okamoto, K.; Aoki, K. Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J. 1963, 27, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Enright, E.F.; Griffin, B.T.; Gahan, C.G.M.; Joyce, S.A. Microbiome-mediated bile acid modification: Role in intestinal drug absorption and metabolism. Pharmacol. Res. 2018, 133, 170–186. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Sun, Y.; Wang, R.; Zhang, J.; Jia, Z. Plateau hypoxia attenuates the metabolic activity of intestinal flora to enhance the bioavailability of nifedipine. Drug Deliv. 2018, 25, 1175–1181. [Google Scholar] [CrossRef]
- Berg, J.; Brandt, K.K.; Al-Soud, W.A.; Holm, P.E.; Hansen, L.H.; Sørensen, S.J.; Nybroe, O. Selection for Cu-tolerant bacterial communities with altered composition, but unaltered richness, via long-term Cu exposure. Appl. Environ. Microbiol. 2012, 78, 7438–7446. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Su, M.; Xie, G.; Ferslew, B.C.; Brouwer, K.L.; Rajani, C.; Liu, C.; Jia, W. Key Role for the 12-Hydroxy Group in the Negative Ion Fragmentation of Unconjugated C24 Bile Acids. Anal. Chem. 2016, 88, 7041–7048. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Shu, N.; Xu, P.; Wang, F.; Zhong, Z.; Sun, B.; Li, F.; Zhang, M.; Zhao, K.; Tang, X.; et al. Involvement of pregnane X receptor in the impaired glucose utilization induced by atorvastatin in hepatocytes. Biochem. Pharmacol. 2016, 100, 98–111. [Google Scholar] [CrossRef]
- Patki, K.C.; Von Moltke, L.L.; Greenblatt, D.J. In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: Role of cyp3a4 and cyp3a5. Drug Metab. Dispos. Biol. Fate Chem. 2003, 31, 938–944. [Google Scholar] [CrossRef]
- von Moltke, L.L.; Greenblatt, D.J.; Harmatz, J.S.; Shader, R.I. Alprazolam metabolism in vitro: Studies of human, monkey, mouse, and rat liver microsomes. Pharmacology 1993, 47, 268–276. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Goldstein, I. Protocol for Primary Mouse Hepatocyte Isolation. STAR Protoc. 2020, 1, 100086. [Google Scholar] [CrossRef]
- Kamareddine, L.; Najjar, H.; Sohail, M.U.; Abdulkader, H.; Al-Asmakh, M. The Microbiota and Gut-Related Disorders: Insights from Animal Models. Cells 2020, 9, 2401. [Google Scholar] [CrossRef]
- Kyoung, J.; Atluri, R.R.; Yang, T. Resistance to Antihypertensive Drugs: Is Gut Microbiota the Missing Link? Hypertension 2022, 79, 2138–2147. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Yang, W.; Yu, H.; He, Q.; Xu, H.; Li, S.; Shang, Z.; Gao, X.; Wang, Y.; et al. Gut Microbiota in Adipose Tissue Dysfunction Induced Cardiovascular Disease: Role as a Metabolic Organ. Front. Endocrinol. 2021, 12, 749125. [Google Scholar] [CrossRef]
- Chen, H.Q.; Gong, J.Y.; Xing, K.; Liu, M.Z.; Ren, H.; Luo, J.Q. Pharmacomicrobiomics: Exploiting the Drug-Microbiota Interactions in Antihypertensive Treatment. Front. Med. 2021, 8, 742394. [Google Scholar] [CrossRef] [PubMed]
- Doestzada, M.; Vila, A.V.; Zhernakova, A.; Koonen, D.P.Y.; Weersma, R.K.; Touw, D.J.; Kuipers, F.; Wijmenga, C.; Fu, J. Pharmacomicrobiomics: A novel route towards personalized medicine? Protein Cell 2018, 9, 432–445. [Google Scholar] [CrossRef]
- Flowers, S.A.; Bhat, S.; Lee, J.C. Potential Implications of Gut Microbiota in Drug Pharmacokinetics and Bioavailability. Pharmacotherapy 2020, 40, 704–712. [Google Scholar] [CrossRef]
- Tuteja, S.; Ferguson, J.F. Gut Microbiome and Response to Cardiovascular Drugs. Circ. Genom. Precis. Med. 2019, 12, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Yu, J.S.; Yoo, H.H.; Kim, D.H. The role of gut microbiota in the pharmacokinetics of antihypertensive drugs. Pharmacol. Res. 2018, 130, 164–171. [Google Scholar] [CrossRef]
- Mishima, E.; Abe, T. Role of the microbiota in hypertension and antihypertensive drug metabolism. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2022, 45, 246–253. [Google Scholar] [CrossRef]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimaraes, V.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Yuasa, H.; Inoue, K.; Iwao, T.; Tanaka, K.; Ooi, K.; Hayashi, Y. Effect of Lactobacillus casei on the absorption of nifedipine from rat small intestine. Drug Metab. Pharmacokinet. 2007, 22, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F. NAFLD: Mechanisms, Treatments, and Biomarkers. Biomolecules 2022, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Saito, N.; Ikarashi, N.; Ito, K.; Yamamoto, M.; Ishige, A.; Watanabe, K.; Sugiyama, K. Intestinal flora induces the expression of Cyp3a in the mouse liver. Xenobiotica 2009, 39, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.T.; Kliewer, S.A. Use of the nuclear receptor PXR to predict drug interactions. Toxicology 2000, 153, 1–10. [Google Scholar] [CrossRef]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Martin, M.V.; Beaune, P.H.; Kremers, P.; Wolff, T.; Waxman, D.J. Characterization of rat and human liver microsomal cytochrome P-450 forms involved in nifedipine oxidation, a prototype for genetic polymorphism in oxidative drug metabolism. J. Biol. Chem. 1986, 261, 5051–5060. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef]
- Schuetz, E.G.; Strom, S.; Yasuda, K.; Lecureur, V.; Assem, M.; Brimer, C.; Lamba, J.; Kim, R.B.; Ramachandran, V.; Komoroski, B.J.; et al. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J. Biol. Chem. 2001, 276, 39411–39418. [Google Scholar] [CrossRef]
- Watkins, R.E.; Wisely, G.B.; Moore, L.B.; Collins, J.L.; Lambert, M.H.; Williams, S.P.; Willson, T.M.; Kliewer, S.A.; Redinbo, M.R. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science 2001, 292, 2329–2333. [Google Scholar] [CrossRef]
- LeCluyse, E.L. Pregnane X receptor: Molecular basis for species differences in CYP3A induction by xenobiotics. Chem.-Biol. Interact. 2001, 134, 283–289. [Google Scholar] [CrossRef]
- Mun, S.J.; Lee, J.; Chung, K.S.; Son, M.Y.; Son, M.J. Effect of Microbial Short-Chain Fatty Acids on CYP3A4-Mediated Metabolic Activation of Human Pluripotent Stem Cell-Derived Liver Organoids. Cells 2021, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Ogura, J.; Sivaprakasam, S.; Ganapathy, V. Gut Microbiome and Colon Cancer: Role of Bacterial Metabolites and Their Molecular Targets in the Host. Curr. Color. Cancer Rep. 2017, 13, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cheng, Y.; Zhang, Y.; Huang, S.; Liu, Z.; Wang, X. Lactobacillus rhamnosus induces CYP3A and changes the pharmacokinetics of verapamil in rats. Toxicol. Lett. 2021, 352, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Q.; Liu, Y.; Wang, L.; Ge, Z.; Li, Z.; Feng, S.; Wu, C. Gut microbiota and hypertension: Association, mechanisms and treatment. Clin. Exp. Hypertens. 2023, 45, 2195135. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Wistar | SHR |
|---|---|---|---|
| AUC0–t | ng·h/mL | 16,755.62 ± 2963.72 | 13,635.22 ± 2666.00 * |
| AUC0–∞ | ng·h/mL | 16,780.48 ± 2977.64 | 14,073.07 ± 2974.92 |
| Cmax | ng/mL | 3165.00 ± 520.52 | 2233.33 ± 417.70 *** |
| Tmax | h | 2.21 ± 0.94 | 1.86 ± 1.74 |
| MRT0–t | h | 3.85 ± 0.34 | 5.09 ± 1.13 *** |
| MRT0–∞ | h | 3.88 ± 0.36 | 5.82 ± 2.48 *** |
| T1/2 | h | 2.15 ± 0.70 | 4.22 ± 1.87 **** |
| Time (h) | Wistar | SHR |
|---|---|---|
| 0.167 | 744.10 ± 245.40 | 771.80 ± 267.10 |
| 0.33 | 1666.00 ± 311.60 | 1462.00 ± 348.90 |
| 0.5 | 2125.00 ± 380.50 | 1771.00 ± 475.50 |
| 0.75 | 2451.00 ± 422.50 | 1748.00 ± 317.00 ** |
| 1 | 2586.00 ± 318.90 | 1969.00 ± 373.00 ** |
| 2.33 | 2881.00 ± 600.70 | 1661.00 ± 521.70 *** |
| 4 | 2440.00 ± 1024.00 | 910.80 ± 331.80 *** |
| 6 | 717.00 ± 272.70 | 988.90 ± 782.10 |
| 8 | 330.20 ± 106.40 | 488.90 ± 177.30 * |
| 24 | 6.11 ± 3.69 | 44.94 ± 67.24 ** |
| Metabolite | Class | Uni_P | Uni_FDR | FC | log2FC | OPLSDA_VIP |
|---|---|---|---|---|---|---|
| GUDCA | Primary BAs | 0.0030 | 0.0204 | 113.81 | 6.83 | 1.5485 |
| GHDCA | Secondary BAs | 0.0032 | 0.0204 | 50.33 | 5.65 | 1.5352 |
| GCA | Primary BAs | 2.06 × 10−4 | 0.0086 | 47.29 | 5.56 | 1.6696 |
| GDCA | Secondary BAs | 0.0039 | 0.0204 | 46.69 | 5.54 | 1.5250 |
| GCDCA | Primary BAs | 0.0089 | 0.0313 | 31.92 | 5.00 | 1.4863 |
| THCA | Secondary BAs | 0.0010 | 0.0139 | 27.41 | 4.78 | 1.6344 |
| TCA | Primary BAs | 4.87 × 10−4 | 0.0102 | 8.84 | 3.14 | 1.5364 |
| UCA | Secondary BAs | 0.0115 | 0.0371 | 7.75 | 2.95 | 1.2438 |
| TDCA | Secondary BAs | 0.0089 | 0.0313 | 3.32 | 1.73 | 1.2045 |
| TCDCA | Primary BAs | 0.0089 | 0.0313 | 2.63 | 1.40 | 1.1256 |
| TαMCA | Primary BAs | 0.0355 | 0.0784 | 2.34 | 1.23 | 1.1112 |
| 6-KetoLCA | Secondary BAs | 0.0232 | 0.0610 | 0.62 | −0.68 | 1.0576 |
| UDCA | Primary BAs | 0.0426 | 0.0895 | 0.50 | −1.01 | 1.0772 |
| muroCA | Secondary BAs | 0.0288 | 0.0712 | 0.42 | −1.24 | 1.1990 |
| βHDCA | Secondary BAs | 0.0089 | 0.0313 | 0.15 | −2.70 | 1.3869 |
| HDCA | Secondary BAs | 0.0015 | 0.0158 | 0.12 | −3.06 | 1.5747 |
| Parameter | Unit | Wistar | SHR |
|---|---|---|---|
| AST | U/L | 432.90 ± 182.80 | 498.20 ± 66.04 |
| ALT | U/L | 99.73 ± 45.88 | 90.33 ± 56.11 |
| TBA | μmol/L | 6.24 ± 2.35 | 8.30 ± 3.43 |
| BUN | mmol/L | 20.31 ± 2.77 | 21.67 ± 2.06 |
| Cr | μmol/L | 58.54 ± 10.28 | 52.78 ± 8.35 |
| UA | μmol/L | 142.20 ± 57.29 | 173.40 ± 72.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, R.; Yang, H.; Zhu, P.; Liu, Y.; Zhang, Y.; Zhang, W.; Zhou, H.; Li, X.; Li, Q. Effect of Gut Microbiota on the Pharmacokinetics of Nifedipine in Spontaneously Hypertensive Rats. Pharmaceutics 2023, 15, 2085. https://doi.org/10.3390/pharmaceutics15082085
Zhou R, Yang H, Zhu P, Liu Y, Zhang Y, Zhang W, Zhou H, Li X, Li Q. Effect of Gut Microbiota on the Pharmacokinetics of Nifedipine in Spontaneously Hypertensive Rats. Pharmaceutics. 2023; 15(8):2085. https://doi.org/10.3390/pharmaceutics15082085
Chicago/Turabian StyleZhou, Rong, Haijun Yang, Peng Zhu, Yujie Liu, Yanjuan Zhang, Wei Zhang, Honghao Zhou, Xiong Li, and Qing Li. 2023. "Effect of Gut Microbiota on the Pharmacokinetics of Nifedipine in Spontaneously Hypertensive Rats" Pharmaceutics 15, no. 8: 2085. https://doi.org/10.3390/pharmaceutics15082085
APA StyleZhou, R., Yang, H., Zhu, P., Liu, Y., Zhang, Y., Zhang, W., Zhou, H., Li, X., & Li, Q. (2023). Effect of Gut Microbiota on the Pharmacokinetics of Nifedipine in Spontaneously Hypertensive Rats. Pharmaceutics, 15(8), 2085. https://doi.org/10.3390/pharmaceutics15082085