

SGLT2 Inhibitors in the Treatment of Diabetic Kidney Disease: More than Just Glucose Regulation


Abstract
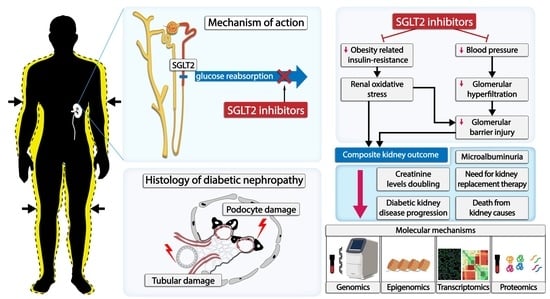
1. Introduction
2. Pathophysiology of CKD in Patients with T2DM
3. SGLT2 Transporters and SGLT2 Inhibitors in the Treatment of Diabetic Kidney Disease

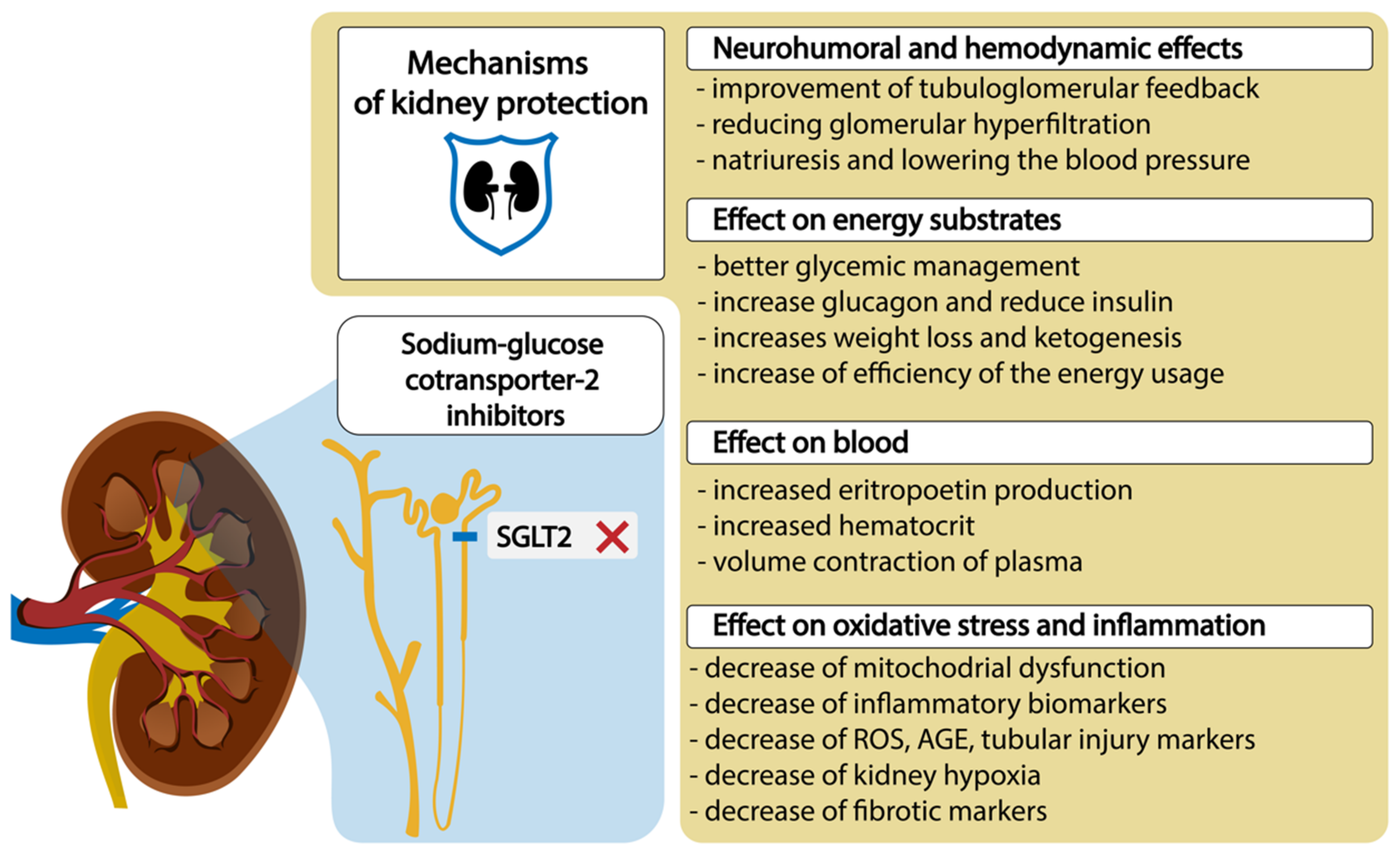
4. Treatment with SGLT2 Inhibitors and Kidney Outcomes

{kind=link}
{kind=link}
{kind=link}
| Study | Study Design | No. of Patients | Drug and Dosage (mg)/Comparator | Median Follow-Up (Months) | eGFR (mL/min/1.73 m2) | UACR (mg/g) | Outcomes | Ref. |
|---|---|---|---|---|---|---|---|---|
| CREDENCE | RCT | 4.401 | canagliflozin 100/placebo | 31.2 | 30–59 | >300 | HR 0.66 (95% CI 0.53–0.81); p < 0.001 for canagliflozin group, indicating a 34% lower relative risk for the composite outcome of end-stage kidney disease; doubling of serum creatinine level or death from kidney causes | [53] |
| CANVAS | RCT | 10.142 | canagliflozin 100/300/placebo | 29 | 30–59 | >300 | HR 0.60 (95% CI 0.47–0.77) for canagliflozin group, indicating a 40% lower relative risk for the composite outcome of end-stage kidney disease; doubling of serum creatinine level or death from kidney causes | [54] |
| DAPA-CKD | RCT | 4.000 | dapagliflozin 10/placebo | 28.8 | 25–45 | >1000 | HR 0.56 (95% CI 0.45–0.68) fort he composite composite of decline in eGFR of ≥50%, ESKD, or death from kidney causes | [58] |
| DAPA-HF | RCT | 4.744 | dapagliflozin 10/placebo | 18.2 | 30–59 | / | HR 0.71 (95% CI 0.44–1.16) for the composite of worsening kidney function, i.e., reduction of ≥50% in the eGFR sustained for ≥28 days; ESKD, or death from kidney causes | [56] |
| DECLARE TIMI-58 | RCT | 17.160 | dapagliflozin 10/placebo | 50.4 | <60 | >300 | HR 0.53 (95% CI 0.43–0.66), p < 0.0001 for dapagliflozin group, indicating 47% lower relative risk for the composite outcome of end-stage kidney disease; doubling of serum creatinine level or death from kidney causes | [55] |
| DELIGHT | RCT | 1.187 | dapagliflozin 10/dapagliflozin 10 + saxagliptin 2.5/placebo | 6 | 25–75 | 30–3500? | The difference in mean UACR change from baseline was −21.0% (95% CI −34.1 to −5.2; p = 0·011) for dapagliflozin and −38.0% (−48.2 to −25.8; p < 0.0001 for dapagliflozin–saxagliptin | [57] |
| EMPA-REG | RCT | 7.020 | empagliflozin 10/25/placebo | 37.2 | 30–59 | >300 | HR 0.61 (95% CI 0.53–0.70), p < 0.001 in empagliflozin group, indicating 39% lower relative risk for the composite outcome of end-stage kidney disease; doubling of serum creatinine level or death from kidney causes | [60] |
| EMPEROR reduced and preserved | RCT | 9.718 | empagliflozin 10/placebo | 16 | 20–59 | >300 | HR 0.50 (95% CI 0.32–0.77) and HR 0.95 (95% CI 0.73–1.24) for the composite kidney outcome of chronic dialysis or kidney transplantation or sustained reduction by ≥40% in eGFR or sustained eGFR | [61,62] |
| EMPA-KIDNEY | RCT | 6.600 | empagliflozin 10/placebo | 24 | 20–45 or 45–90 | ≥200 | HR 0.72 (95% CI 0.64 to 0.82; p < 0.001) for end-stage kidney disease (ESKD), a sustained decline in eGFR to less than 10 mL per minute per 1.73 m2; a sustained decline in eGFR of at least 40% from baseline, or death from kidney causes | [59] |
| VERTIS-CV | RCT | 8.246 | ertugliflozin 15/5/placebo | 42 | 30–59 | >300 | HR 0.81 (95% CI 0.63–1.04) in composite kidney outcome: doubling of serum creatinine, kidney replacement therapy, or death from kidney causes | [64] |
| SCORED | RCT | 10.584 | sotagliflozin 200 or 400/placebo | 16/15.9 | 25–59 | >300 | HR 0.71 (95% CI 0.46–1.08) for the composite outcome of first occurrence of sustained decrease of ≥50% in eGFR from baseline for ≥30 days, long-term dialysis, kidney transplantation, or sustained eGFR 15 mL/min/1.73 m2 for ≥ 30 days | [65] |
5. Molecular Mechanisms and Molecular Genetics of SGLT Action—Potential Biomarkers of Treatment
5.1. Genetic and Genomic Factors Related to SGLT2 Action
5.2. Epigenomic Factors Related to CKD and SGLT2 Action
5.3. Proteomic and Inflammatory Biomarkers
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeFronzo, R.A.; Reeves, W.B.; Awad, A.S. Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 2021, 17, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; de Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic kidney disease: A report from an ADA Consensus Conference. Diabetes Care 2014, 37, 2864–2883. [Google Scholar] [CrossRef]
- National Kidney Foundation. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- Faselis, C.; Katsimardou, A.; Imprialos, K.; Deligkaris, P.; Kallistratos, M.S.; Dimitriadis, K. Microvascular Complications of Type 2 Diabetes Mellitus. Curr. Vasc. Pharmacol. 2020, 18, 117–124. [Google Scholar] [CrossRef]
- Crasto, W.; Patel, V.; Davies, M.J.; Khunti, K. Prevention of Microvascular Complications of Diabetes. Endocrinol. Metab. Clin. North Am. 2021, 50, 431–455. [Google Scholar] [CrossRef] [PubMed]
- Nitta, K.; Goto, S.; Masakane, I.; Hanafusa, N.; Taniguchi, M.; Hasegawa, T.; Nakai, S.; Wada, A.; Hamano, T.; Hoshino, J.; et al. Annual dialysis data report for 2017, JSDT Renal Data Registry: Survey methods, facility data, incidence, prevalence, and mortality. Ren. Replace. Ther. 2020, 6, 41. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 6. Glycemic Targets: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45 (Suppl. S1), S83–S96. [Google Scholar]
- Nathan, D.M.; DCCT/EDIC Research Group. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: Overview. Diabetes Care 2014, 37, 9–16. [Google Scholar] [CrossRef]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 11. Chronic Kidney Disease and Risk Management: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45 (Suppl. S1), S175–S184. [Google Scholar] [CrossRef]
- Sasso, F.C.; Simeon, V.; Galiero, R.; Caturano, A.; De Nicola, L.; Chiodini, P.; Rinaldi, L.; Salvatore, T.; Lettieri, M.; Nevola, R.; et al. The number of risk factors not at target is associated with cardiovascular risk in a type 2 diabetic population with albuminuria in primary cardiovascular prevention. Post-hoc analysis of the NID-2 trial. Cardiovasc. Diabetol. 2022, 21, 235. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.G.; Preiss, D.; Haynes, R.; von Eynatten, M.; Staplin, N.; Hauske, S.J.; George, J.T.; Green, J.B.; Landray, M.J.; Baigent, C.; et al. The potential for improving cardio-renal outcomes by sodium-glucose co-transporter-2 inhibition in people with chronic kidney disease: A rationale for the EMPA-KIDNEY study. Clin. Kidney J. 2018, 11, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Mima, A. Inflammation and Oxidative Stress in Diabetic Nephropathy: New Insights on Its Inhibition as New Therapeutic Targets. J. Diabetes Res. 2013, 2013, 248563. [Google Scholar] [CrossRef]
- Salvatore, T.; Galiero, R.; Caturano, A.; Rinaldi, L.; Di Martino, A.; Albanese, G.; Di Salvo, J.; Epifani, R.; Marfella, R.; Docimo, G.; et al. An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 3651. [Google Scholar] [CrossRef] [PubMed]
- Van Bommel, E.J.; Muskiet, M.H.; van Baar, M.J.; Tonneijck, L.; Smits, M.M.; Emanuel, A.L.; Bozovic, A.; Danser, A.J.; Geurts, F.; Hoorn, E.J.; et al. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int. 2020, 97, 202–212. [Google Scholar] [CrossRef]
- Ott, C.; Jung, S.; Korn, M.; Kannenkeril, D.; Bosch, A.; Kolwelter, J.; Striepe, K.; Bramlage, P.; Schiffer, M.; Schmieder, R.E. Renal hemodynamic effects differ between antidiabetic combination strategies: Randomized controlled clinical trial comparing empagliflozin/linagliptin with metformin/insulin glargine. Cardiovasc. Diabetol. 2021, 20, 178. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.I.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; von Eynatten, M.; Wanner, C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: An exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 610–621. [Google Scholar] [CrossRef]
- Lachaux, M.; Soulié, M.; Hamzaoui, M.; Bailly, A.; Nicol, L.; Rémy-Jouet, I.; Renet, S.; Vendeville, C.; Gluais-Dagorn, P.; Hallakou-Bozec, S.; et al. Short-and long-term administration of imeglimin counters cardiorenal dysfunction in a rat model of metabolic syndrome. Endocrinol. Diabetes Metab. 2020, 3, e00128. [Google Scholar] [CrossRef]
- Kao, M.P.C.; Ang, D.S.C.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef]
- Krishan, P.; Chakkarwar, V.A. Diabetic nephropathy: Aggressive involvement of oxidative stress. J. Pharm. Edu. Res. 2011, 2, 35–41. [Google Scholar]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Khazim, K.; Gorin, Y.; Cavaglieri, R.C.; Abboud, H.E.; Fanti, P. The antioxidant silybin prevents high glucose-induced oxidative stress and podocyte injury in vitro and in vivo. Am. J. Physiol. Physiol. 2013, 305, F691–F700. [Google Scholar] [CrossRef] [PubMed]
- Samsu, N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. BioMed Res. Int. 2021, 2021, 1497449. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Krum, H.; Wilkinson-Berka, J.; Kelly, D.J. The renin-angiotensin system and the long-term complications of diabetes: Pathophysiological and therapeutic considerations. Diabet. Med. 2003, 20, 607–621. [Google Scholar] [CrossRef]
- Shang, J.; Wang, L.; Zhang, Y.; Zhang, S.; Ning, L.; Zhao, J.; Cheng, G.; Liu, D.; Xiao, J.; Zhao, Z. Chemerin/ChemR23 axis promotes inflammation of glomerular endothelial cells in diabetic nephropathy. J. Cell. Mol. Med. 2019, 23, 3417–3428. [Google Scholar] [CrossRef]
- Nakamura, A.; Shikata, K.; Hiramatsu, M.; Nakatou, T.; Kitamura, T.; Wada, J.; Itoshima, T.; Makino, H. Serum Interleukin-18 Levels Are Associated With Nephropathy and Atherosclerosis in Japanese Patients With Type 2 Diabetes. Diabetes Care 2005, 28, 2890–2895. [Google Scholar] [CrossRef]
- Saraheimo, M.; Teppo, A.-M.; Forsblom, C.; Fagerudd, J.; Groop, P.-H. Diabetic nephropathy is associated with low-grade inflammation in Type 1 diabetic patients. Diabetologia 2003, 46, 1402–1407. [Google Scholar] [CrossRef]
- Moriwaki, Y.; Yamamoto, T.; Shibutani, Y.; Aoki, E.; Tsutsumi, Z.; Takahashi, S.; Okamura, H.; Koga, M.; Fukuchi, M.; Hada, T. Elevated levels of interleukin-18 and tumor necrosis factor-α in serum of patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy. Metabolism 2003, 52, 605–608. [Google Scholar] [CrossRef]
- Tavafi, M. Diabetic nephropathy and antioxidants. J. Nephropathol. 2013, 2, 20–27. [Google Scholar] [CrossRef]
- Pérez-Morales, R.E.; del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Inflammation in Diabetic Kidney Disease. Nephron 2019, 143, 12–16. [Google Scholar] [CrossRef]
- Menne, J.; Park, J.-K.; Boehne, M.; Elger, M.; Lindschau, C.; Kirsch, T.; Meier, M.; Gueler, F.; Fiebeler, A.; Bahlmann, F.H.; et al. Diminished Loss of Proteoglycans and Lack of Albuminuria in Protein Kinase C-α—Deficient Diabetic Mice. Diabetes 2004, 53, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Castellani, P.; Balza, E.; Rubartelli, A. Inflammation, DAMPs, Tumor Development, and Progression: A Vicious Circle Orchestrated by Redox Signaling. Antioxid. Redox Signal. 2014, 20, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Fukui, M.; Ebihara, I.; Osada, S.; Nagaoka, I.; Tomino, Y.; Koide, H. mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes 1993, 42, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.F.; Fang, R.Y.; Hsieh, H.L.; Chi, P.L.; Lin, C.C.; Hsiao, L.D.; Wu, C.C.; Wang, J.S.; Yang, C.M. Involvement of MAPKs and NF-kappaB in tumor necrosis factor alpha-induced vascular cell adhesion molecule 1 expression in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2010, 62, 105–116. [Google Scholar] [CrossRef]
- Deng, D.; Yan, N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016, 25, 546–558. [Google Scholar] [CrossRef]
- Zaccardi, F.; Webb, D.R.; Htike, Z.Z.; Youssef, D.; Khunti, K.; Davies, M.J. Efficacy and safety of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes mellitus: Systematic review and network meta-analysis. Diabetes Obes. Metab. 2016, 18, 783–794. [Google Scholar] [CrossRef]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Har, R.; Fagan, N.; Johansen, O.; Woerle, H.-J.; von Eynatten, M.; Broedl, U.C. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 28. [Google Scholar] [CrossRef]
- Lee, P.C.; Ganguly, S.; Goh, S.-Y. Weight loss associated with sodium-glucose cotransporter-2 inhibition: A review of evidence and underlying mechanisms. Obes. Rev. 2018, 19, 1630–1641. [Google Scholar] [CrossRef]
- Bolinder, J.; Ljunggren, Ö.; Kullberg, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sugg, J.; Parikh, S. Effects of Dapagliflozin on Body Weight, Total Fat Mass, and Regional Adipose Tissue Distribution in Patients with Type 2 Diabetes Mellitus with Inadequate Glycemic Control on Metformin. J. Clin. Endocrinol. Metab. 2012, 97, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Schork, A.; Saynisch, J.; Vosseler, A.; Jaghutriz, B.A.; Heyne, N.; Peter, A.; Häring, H.U.; Stefan, N.; Fritsche, A.; Artunc, F. Effect of SGLT2 inhibitors on body composition, fluid status and renin-angiotensin-aldosterone system in type 2 diabetes: A prospective study using bioimpedance spectroscopy. Cardiovasc. Diabetol. 2019, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sridhar, V.S.; Boulet, J.; Dharia, A.; Khan, A.; Lawler, P.R.; Cherney, D.Z. Cardiorenal protection with SGLT2 inhibitors in patients with diabetes mellitus: From biomarkers to clinical outcomes in heart failure and diabetic kidney disease. Metabolism 2022, 126, 154918. [Google Scholar] [CrossRef]
- Liu, H.; Sridhar, V.S.; Montemayor, D.; Lovblom, L.E.; Lytvyn, Y.; Ye, H.; Kim, J.; Ali, M.T.; Scarr, D.; Lawler, P.R.; et al. Changes in plasma and urine metabolites associated with empagliflozin in patients with type 1 diabetes. Diabetes Obes. Metab. 2021, 23, 2466–2475. [Google Scholar] [CrossRef]
- Dharia, A.; Khan, A.; Sridhar, V.S.; Cherney, D.Z. SGLT2 Inhibitors: The Sweet Success for Kidneys. Annu. Rev. Med. 2023, 74, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Kuno, A.; Tanno, M.; Sato, T.; Ohno, K.; Shibata, S.; Nakata, K.; Sugawara, H.; Abe, K.; Igaki, Y.; et al. Canagliflozin, a sodium–glucose cotransporter 2 inhibitor, normalizes renal susceptibility to type 1 cardiorenal syndrome through reduction of renal oxidative stress in diabetic rats. J. Diabetes Investig. 2019, 10, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Alshnbari, A.S.; Millar, S.A.; O’sullivan, S.E.; Idris, I. Effect of Sodium-Glucose Cotransporter-2 Inhibitors on Endothelial Function: A Systematic Review of Preclinical Studies. Diabetes Ther. 2020, 11, 1947–1963. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.-J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef]
- Liu, H.; Sridhar, V.S.; Lovblom, L.E.; Lytvyn, Y.; Burger, D.; Burns, K.; Brinc, D.; Lawler, P.R.; Cherney, D.Z. Markers of Kidney Injury, Inflammation, and Fibrosis Associated With Ertugliflozin in Patients With CKD and Diabetes. Kidney Int. Rep. 2021, 6, 2095–2104. [Google Scholar] [CrossRef]
- Mima, A. Mitochondria-targeted drugs for diabetic kidney disease. Heliyon 2022, 8, e08878. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Kosiborod, M.; Inzucchi, S.E.; Cherney, D.Z. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors. Kidney Int. 2018, 94, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Hesp, A.C.; Schaub, J.A.; Prasad, P.V.; Vallon, V.; Laverman, G.D.; Bjornstad, P.; van Raalte, D.H. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: A promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 2020, 98, 579–589. [Google Scholar] [CrossRef]
- Yu, S.M.-W.; Bonventre, J.V. Acute Kidney Injury and Progression of Diabetic Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Day, C.; Bellary, S. Renal Protection with SGLT2 Inhibitors: Effects in Acute and Chronic Kidney Disease. Curr. Diabetes Rep. 2022, 22, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Shaw, W.; Barrett, T.D.; Weidner-Wells, M.; Deng, H.; Matthews, D.R.; et al. Canagliflozin and renal outcomes in type 2 diabetes: Results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol. 2018, 6, 691–704. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Pollock, C.; Stefánsson, B.; Reyner, D.; Rossing, P.; Sjöström, C.D.; Wheeler, D.C.; Langkilde, A.M.; Heerspink, H.J.L. Albuminuria-lowering effect of dapagliflozin alone and in combination with saxagliptin and effect of dapagliflozin and saxagliptin on glycaemic control in patients with type 2 diabetes and chronic kidney disease (DELIGHT): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 429–441. [Google Scholar]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. DAPA-CKD Trial Committees and Investigators. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- The EMPA-KIDNEY Collaborative Group. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. EMPA-REG OUTCOME Investigators. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Packer, M.; Butler, J.; Zannad, F.; Pocock, S.J.; Filippatos, G.; Ferreira, J.P.; Brueckmann, M.; Jamal, W.; Zeller, C.; Wanner, C.; et al. Empagliflozin and Major Renal Outcomes in Heart Failure. N. Engl. J. Med. 2021, 385, 1531–1533. [Google Scholar] [CrossRef]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in Patients with Diabetes and Chronic Kidney Disease. N. Engl. J. Med. 2021, 384, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Nuffield Department of Population Health Renal Studies Group; SGLT2 inhibitor Meta-Analysis Cardio-Renal Trialists’ Consortium. Impact of diabetes on the effects of sodium glucose co-transporter-2 inhibitors on kidney outcomes: Collaborative meta-analysis of large placebo-controlled trials. Lancet 2022, 400, 1788–1801. [Google Scholar] [CrossRef]
- Georgianos, P.I.; Vaios, V.; Eleftheriadis, T.; Papachristou, E.; Liakopoulos, V. Therapeutic Advances in Diabetic Kidney Disease. Int. J. Mol. Sci. 2023, 24, 2803. [Google Scholar] [CrossRef]
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of Diabetic Kidney Disease: Current and Future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Santer, R.; Calado, J. Familial renal glucosuria and SGLT2: From a mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol. CJASN 2010, 5, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Enigk, U.; Breitfeld, J.; Schleinitz, D.; Dietrich, K.; Halbritter, J.; Fischer-Rosinsky, A.; Enigk, B.; Müller, I.; Spranger, J.; Pfeiffer, A.; et al. Role of genetic variation in the human sodium–glucose cotransporter 2 gene (SGLT2) in glucose homeostasis. Pharmacogenomics 2011, 12, 1119–1126. [Google Scholar] [CrossRef]
- Klen, J.; Goričar, K.; Dolžan, V. Genetic variability in sodium-glucose cotransporter 2 influences glycemic control and risk for diabetic retinopathy in type 2 diabetes patients. J. Med. Biochem. 2020, 39, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Zimdahl, H.; Haupt, A.; Brendel, M.; Bour, L.; Machicao, F.; Salsali, A.; Broedl, U.C.; Woerle, H.-J.; Häring, H.-U.; Staiger, H. Influence of common polymorphisms in the SLC5A2 gene on metabolic traits in subjects at increased risk of diabetes and on response to empagliflozin treatment in patients with diabetes. Pharmacogenet. Genom. 2017, 27, 135–142. [Google Scholar] [CrossRef]
- Shojima, N.; Yamauchi, T. Progress in genetics of type 2 diabetes and diabetic complications. J. Diabetes Investig. 2023, 14, 503–515. [Google Scholar] [CrossRef]
- The Diabetes Control and Complications Trial Research Group. Clustering of long-term complications in families with diabetes in the diabetes control and complications trial. Diabetes 1997, 46, 1829. [Google Scholar] [CrossRef]
- Fox, C.S.; Yang, Q.; Cupples, L.A.; Guo, C.Y.; Larson, M.G.; Leip, E.P.; Wilson, P.W.; Levy, D. Genomewide linkage analysis to serum creatinine, GFR, and creatinine clearance in a community-based population: The Framingham Heart Study. J. Am. Soc. Nephrol. 2004, 15, 2457–2461. [Google Scholar] [CrossRef]
- Cole, J.B.; Florez, J.C. Genetics of diabetes mellitus and diabetes complications. Nat. Rev. Nephrol. 2020, 16, 377–390. [Google Scholar] [CrossRef]
- Available online: https://www.ebi.ac.uk/gwas/efotraits/EFO_0000401 (accessed on 1 June 2023).
- Iyengar, S.K.; Sedor, J.R.; Freedman, B.I.; Kao, W.H.L.; Kretzler, M.; Keller, B.J.; Abboud, H.E.; Adler, S.G.; Best, L.G.; Bowden, D.W.; et al. Genome-Wide Association and Trans-ethnic Meta-Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet. 2015, 11, e1005352. [Google Scholar] [CrossRef]
- Ustinova, M.; Peculis, R.; Rescenko, R.; Rovite, V.; Zaharenko, L.; Elbere, I.; Silamikele, L.; Konrade, I.; Sokolovska, J.; Pirags, V.; et al. Novel susceptibility loci identified in a genome-wide association study of type 2 diabetes complications in population of Latvia. BMC Med. Genom. 2021, 14, 18. [Google Scholar] [CrossRef]
- Mansour, A.; Mousa, M.; Abdelmannan, D.; Tay, G.; Hassoun, A.; Alsafar, H. Microvascular and macrovascular complications of type 2 diabetes mellitus: Exome wide association analyses. Front. Endocrinol. 2023, 14, 143067. [Google Scholar] [CrossRef] [PubMed]
- Kullo, I.J.; Lewis, C.M.; Inouye, M.; Martin, A.R.; Ripatti, S.; Chatterjee, N. Polygenic scores in biomedical research. Nat. Rev. Genet. 2022, 23, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Haloui, M.; Attaoua, R.; Tahir, R.; Hishmih, C.; Harvey, F.; Marois-Blanchet, F.C.; Long, C.; Simon, P.; Santucci, L.; et al. Polygenic risk scores predict diabetes complications and their response to intensive blood pressure and glucose control. Diabetologia 2021, 64, 2012–2025. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef]
- Zuk, O.; Schaffner, S.F.; Samocha, K.; Do, R.; Hechter, E.; Kathiresan, S.; Daly, M.J.; Neale, B.M.; Sunyaev, S.R.; Lander, E.S. Searching for missing heritability: Designing rare variant association studies. Proc. Natl. Acad. Sci. USA 2014, 111, E455–E464. [Google Scholar] [CrossRef]
- Li, H.; Mai, Z.; Yu, S.; Wang, B.; Lai, W.; Chen, G.; Zhou, C.; Liu, J.; Yang, Y.; Chen, S.; et al. Exploring the Pleiotropic Genes and Therapeutic Targets Associated with Heart Failure and Chronic Kidney Disease by Integrating metaCCA and SGLT2 Inhibitors’ Target Prediction. BioMed Res. Int. 2021, 2021, 4229194. [Google Scholar] [CrossRef]
- Chen, Z.; Miao, F.; Paterson, A.D.; Lachin, J.M.; Zhang, L.; Schones, D.E.; Wu, X.; Wang, J.; Tompkins, J.D.; Genuth, S.; et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc. Natl. Acad. Sci. USA 2016, 113, E3002–E3011. [Google Scholar] [CrossRef]
- Marumo, T.; Yagi, S.; Kawarazaki, W.; Nishimoto, M.; Ayuzawa, N.; Watanabe, A.; Ueda, K.; Hirahashi, J.; Hishikawa, K.; Sakurai, H.; et al. Diabetes Induces Aberrant DNA Methylation in the Proximal Tubules of the Kidney. J. Am. Soc. Nephrol. 2015, 26, 2388–2397. [Google Scholar] [CrossRef]
- Ko, Y.A.; Mohtat, D.; Suzuki, M.; Park, A.S.; Izquierdo, M.C.; Han, S.Y.; Kang, H.M.; Sillman, J.M.; Fazzari, M.; Verma, A.; et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013, 14, R108. [Google Scholar] [CrossRef] [PubMed]
- Fraszczyk, E.; Spijkerman, A.M.W.; Zhang, Y.; Brandmaier, S.; Day, F.R.; Zhou, L.; Wackers, P.; Dollé, M.E.T.; Bloks, V.W.; Gào, X.; et al. Epigenome-wide association study of incident type 2 diabetes: A meta-analysis of five prospective European cohorts. Diabetologia 2022, 65, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, W.; Zhu, Y.; Zhang, S.; Jiang, M.; Hu, J.; Zhang, H.-H. Genomic DNA Methylation in Diabetic Chronic Complications in Patients With Type 2 Diabetes Mellitus. Front. Endocrinol. 2022, 13, 896511. [Google Scholar] [CrossRef] [PubMed]
- Giri, A.K.; Prasad, G.; Parekkat, V.; Rajashekar, D.; Tandon, N.; Bharadwaj, D. Epigenome-wide methylation study identified 2 novel CpGs associated with T2DM risk and a network of co-methylated CpGs capable of patient’s classifications. Hum. Mol. Genet. 2023, ddad084. [Google Scholar] [CrossRef]
- Heerbothm, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of Epigenetic Drugs in Disease: An Overview. Genet. Epigenetics 2014, 6, 9–19. [Google Scholar] [CrossRef]
- Gorica, E.; Mohammed, S.A.; Ambrosini, S.; Calderone, V.; Costantino, S.; Paneni, F. Epi-Drugs in Heart Failure. Front. Cardiovasc. Med. 2022, 9, 923014. [Google Scholar] [CrossRef]
- Martinez-Moreno, J.M.; Fontecha-Barriuso, M.; Martin-Sanchez, D.; Guerrero-Mauvecin, J.; Goma-Garces, E.; Fernandez-Fernandez, B.; Carriazo, S.; Sanchez-Niño, M.D.; Ramos, A.M.; Ruiz-Ortega, M.; et al. Epigenetic Modifiers as Potential Therapeutic Targets in Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 4113. [Google Scholar] [CrossRef]
- Zhou, T.; Cheng, X.; He, Y.; Xie, Y.; Xu, F.; Xu, Y.; Huang, W. Function and mechanism of histone β-hydroxybutyrylation in health and disease. Front. Immunol. 2022, 13, 981285. [Google Scholar] [CrossRef]
- Nishitani, S.; Fukuhara, A.; Shin, J.; Okuno, Y.; Otsuki, M.; Shimomura, I. Metabolomic and microarray analyses of adipose tissue of dapagliflozin-treated mice, and effects of 3-hydroxybutyrate on induction of adiponectin in adipocytes. Sci. Rep. 2018, 8, 8805. [Google Scholar] [CrossRef]
- Goričar, K.; Dolžan, V.; Lenassi, M. Extracellular Vesicles: A Novel Tool Facilitating Personalized Medicine and Pharmacogenomics in Oncology. Front. Pharmacol. 2021, 12, 671298. [Google Scholar] [CrossRef]
- Kato, M.; Natarajan, R. MicroRNAs in diabetic nephropathy: Functions, biomarkers, and therapeutic targets. Ann. N. Y. Acad. Sci. 2015, 1353, 72–88. [Google Scholar] [CrossRef] [PubMed]
- Florijn, B.W.; Duijs, J.M.G.J.; Levels, J.H.; Dallinga-Thie, G.M.; Wang, Y.; Boing, A.N.; Yuana, Y.; Stam, W.; Limpens, R.W.A.L.; Au, Y.W.; et al. Diabetic Nephropathy Alters the Distribution of Circulating Angiogenic MicroRNAs Among Extracellular Vesicles, HDL, and Ago-2. Diabetes 2019, 68, 2287–2300. [Google Scholar] [CrossRef]
- Prattichizzo, F.; Matacchione, G.; Giuliani, A.; Sabbatinelli, J.; Olivieri, F.; de Candia, P.; De Nigris, V.; Ceriello, A. Extracellular vesicle-shuttled miRNAs: A critical appraisal of their potential as nano-diagnostics and nano-therapeutics in type 2 diabetes mellitus and its cardiovascular complications. Theranostics 2021, 11, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Seghieri, M.; Giannini, L.; Biancalana, E.; Parolini, F.; Rossi, C.; Dardano, A.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. The Effects of Dapagliflozin on Systemic and Renal Vascular Function Display an Epigenetic Signature. J. Clin. Endocrinol. Metab. 2019, 104, 4253–4263. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Sato, E.; Mishima, E.; Miyazaki, M.; Tanaka, T. What’s New in the Molecular Mechanisms of Diabetic Kidney Disease: Recent Advances. Int. J. Mol. Sci. 2023, 24, 570. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G.; Heerspink, H.J.; Aschauer, C.; Heinzel, A.; Heinze, G.; Kainz, A.; Sunzenauer, J.; Perco, P.; de Zeeuw, D.; Rossing, P.; et al. Systems Biology–Derived Biomarkers to Predict Progression of Renal Function Decline in Type 2 Diabetes. Diabetes Care 2017, 40, 391–397. [Google Scholar] [CrossRef]
- Cefalu, W.T.; Leiter, L.A.; Yoon, K.-H.; Arias, P.; Niskanen, L.; Xie, J.; Balis, D.A.; Canovatchel, W.; Meininger, G. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet 2013, 382, 941–950. [Google Scholar] [CrossRef]
- Sen, T.; Li, J.; Neuen, B.L.; Neal, B.; Arnott, C.; Parikh, C.R.; Coca, S.G.; Perkovic, V.; Mahaffey, K.W.; Yavin, Y.; et al. Effects of the SGLT2 inhibitor canagliflozin on plasma biomarkers TNFR-1, TNFR-2 and KIM-1 in the CANVAS trial. Diabetologia 2021, 64, 2147–2158. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; Micheloni, S.; La Sala, L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is lowgrade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.-G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Liu, X.; Yin, J.; Wu, H.; Cai, X.; Wang, N.; Qian, Y.; Wang, F. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism 2018, 83, 18–24. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klen, J.; Dolžan, V. SGLT2 Inhibitors in the Treatment of Diabetic Kidney Disease: More than Just Glucose Regulation. Pharmaceutics 2023, 15, 1995. https://doi.org/10.3390/pharmaceutics15071995
Klen J, Dolžan V. SGLT2 Inhibitors in the Treatment of Diabetic Kidney Disease: More than Just Glucose Regulation. Pharmaceutics. 2023; 15(7):1995. https://doi.org/10.3390/pharmaceutics15071995
Chicago/Turabian StyleKlen, Jasna, and Vita Dolžan. 2023. "SGLT2 Inhibitors in the Treatment of Diabetic Kidney Disease: More than Just Glucose Regulation" Pharmaceutics 15, no. 7: 1995. https://doi.org/10.3390/pharmaceutics15071995
APA StyleKlen, J., & Dolžan, V. (2023). SGLT2 Inhibitors in the Treatment of Diabetic Kidney Disease: More than Just Glucose Regulation. Pharmaceutics, 15(7), 1995. https://doi.org/10.3390/pharmaceutics15071995