
Rational Design of Topical Semi-Solid Dosage Forms-How Far Are We?


Abstract
1. Introduction
2. Status-Quo of Semi-Solid Formulations in Recently Approved Products
3. Formulation Design
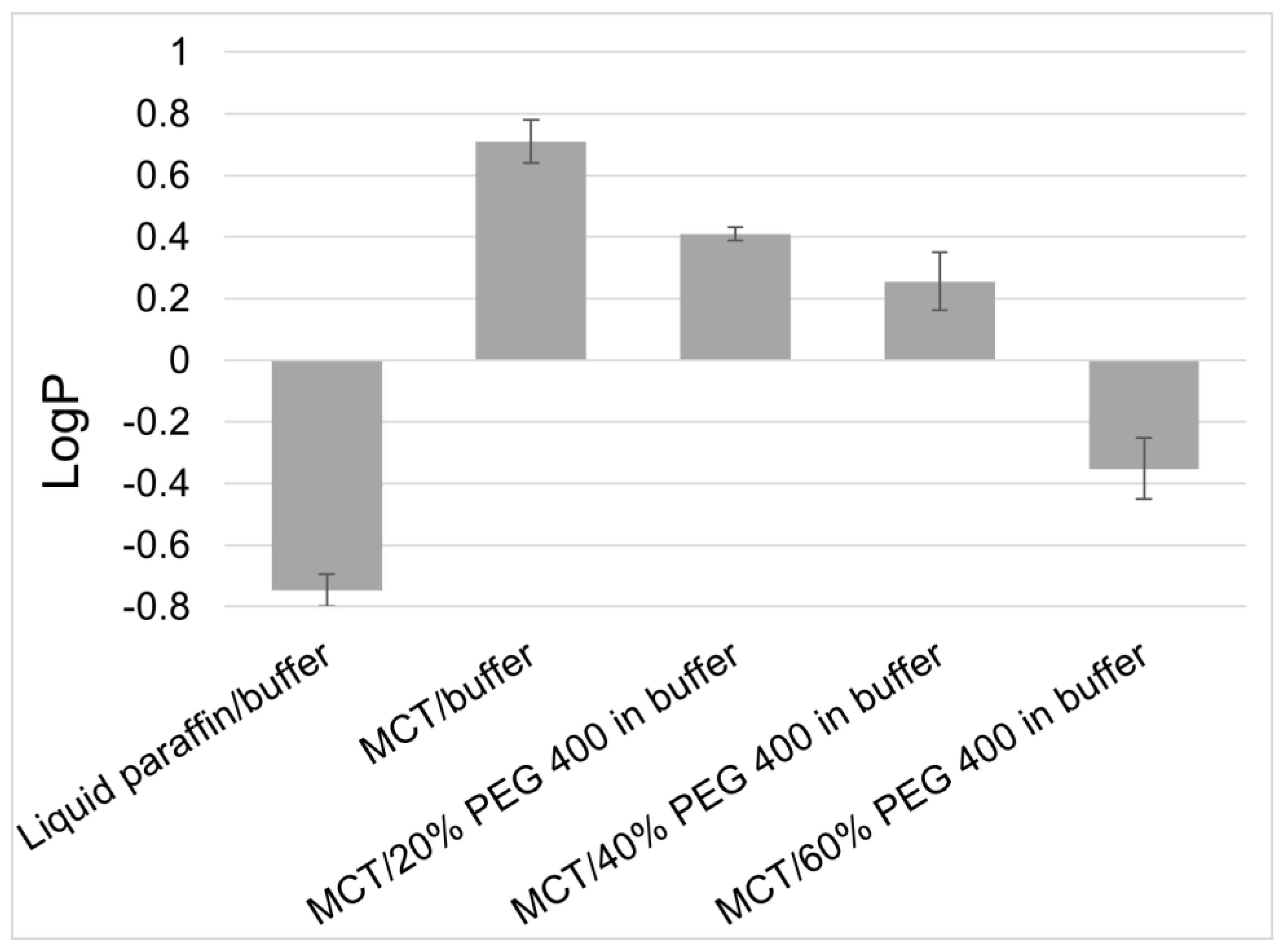
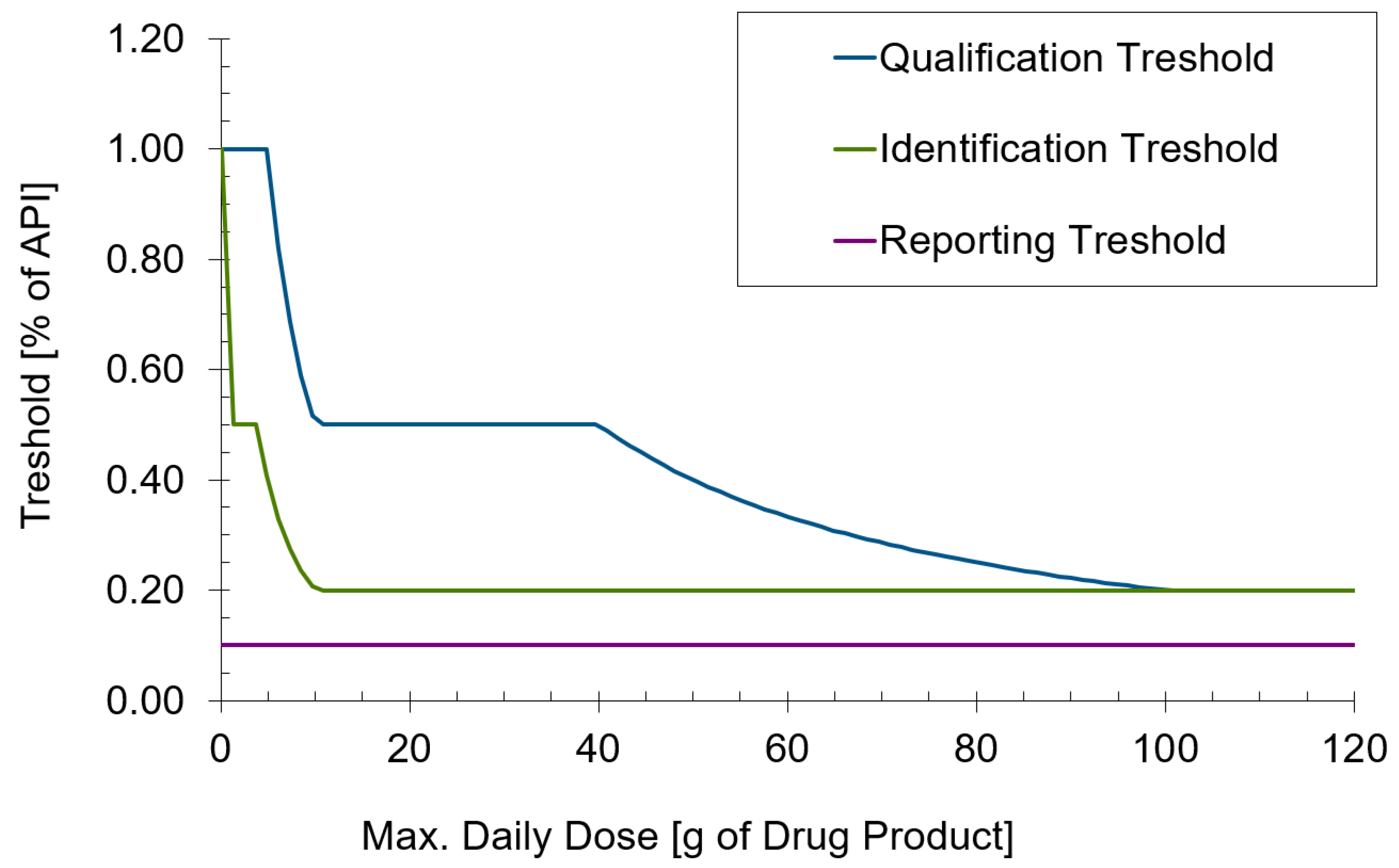
3.1. Selection and Formulatability Assessment of Active Ingredients
3.2. Rational Formulation Design Approaches
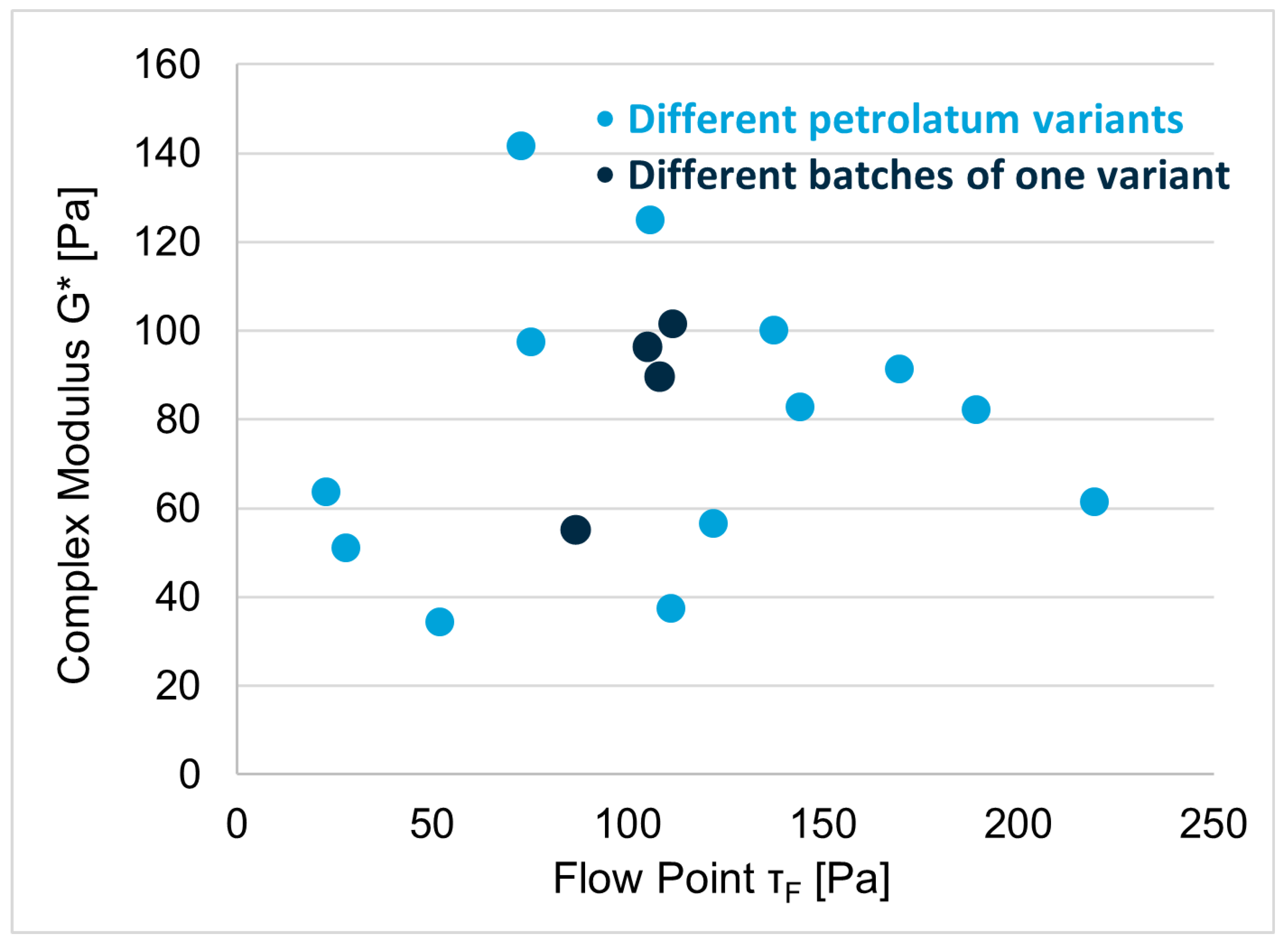
3.3. Excipient Characterization
4. Characterization of Semi-Solid Formulations
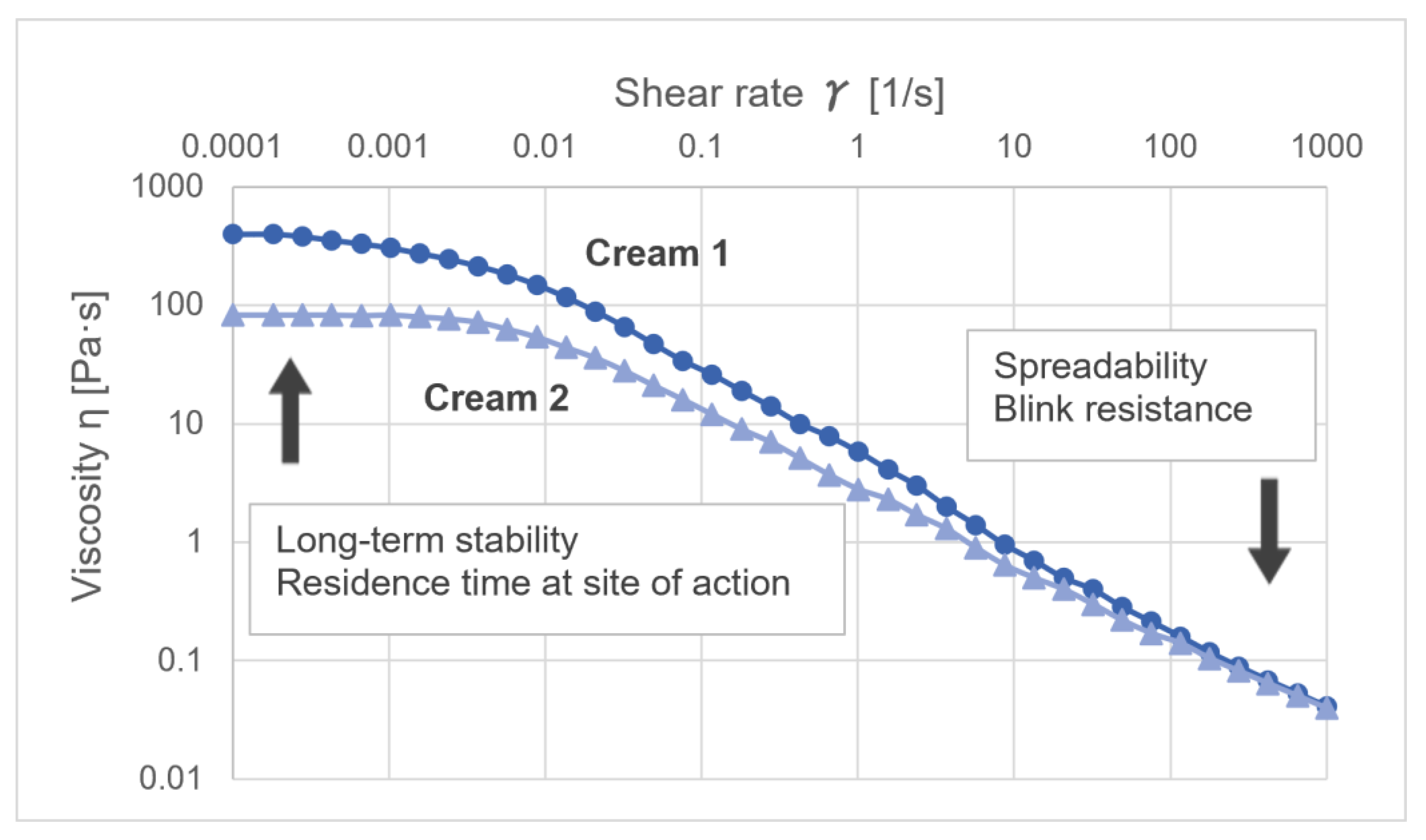
4.1. Rheological Characterization of Semi-Solids
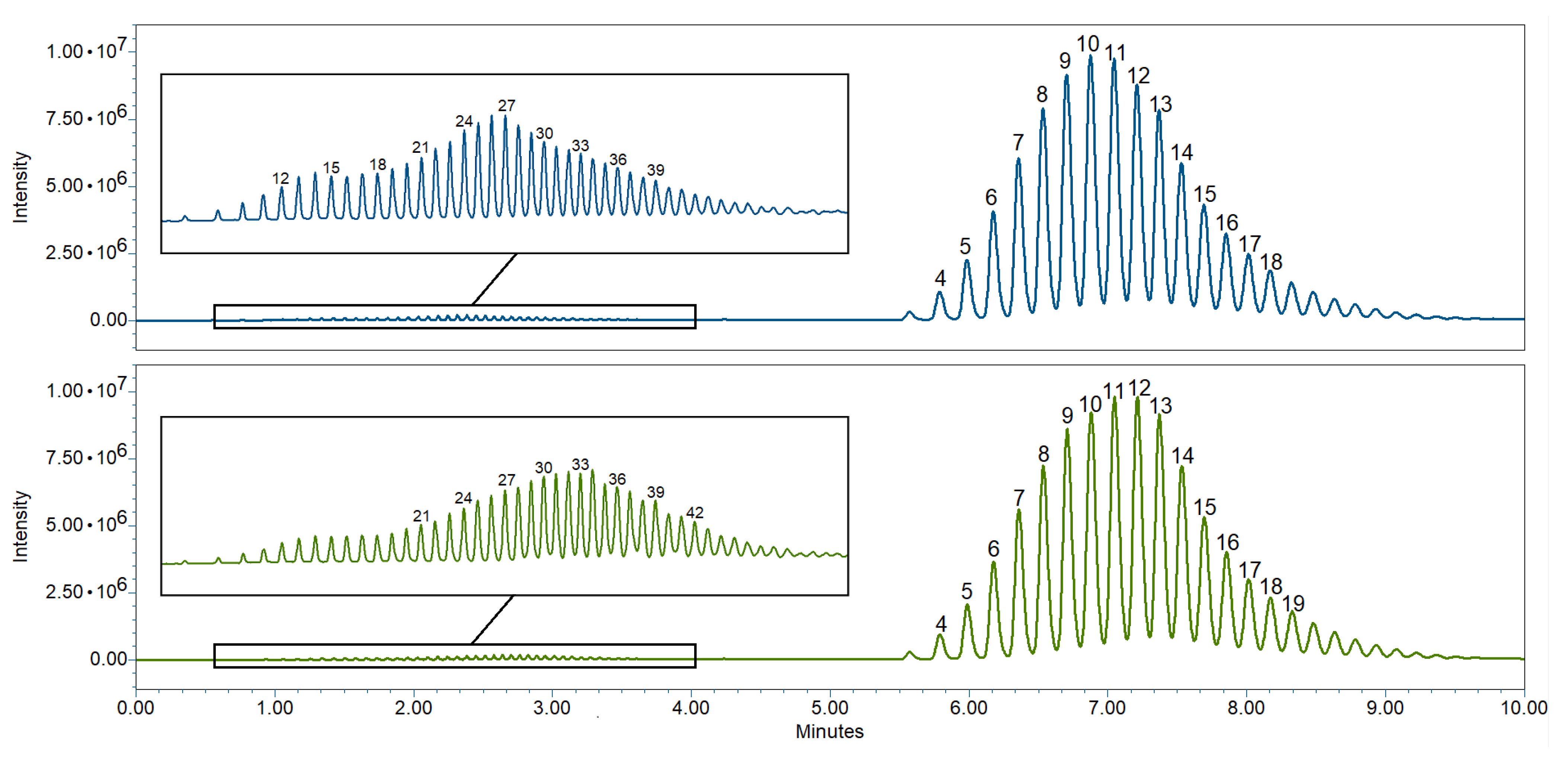
4.2. Chemical Analytics of Semi-Solids
4.3. In Vitro Performance Testing
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Scherphof, G.L.; Fahr, A. Voigt’s Pharmaceutical Technology; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Walters, K.A.; Roberts, M.S. Dermatologic, Cosmeceutic, and Cosmetic Development Therapeutic and Novel Approaches; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Herbig, M.E. Topical Drug Delivery and the Role of Excipients. Chim. Oggi Chem. Today 2022, 40, 34–37. [Google Scholar]
- Dragicevic, N.; Maibach, H.I. (Eds.) Percutaneous Absorption: Drugs, Cosmetics, Mechanisms, Methods, 5th ed.; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar]
- Grégoire, S.; Ribaud, C.; Benech, F.; Meunier, J.R.; Garrigues-Mazert, A.; Guy, R.H. Prediction of chemical absorption into and through the skin from cosmetic and dermatological formulations. Br. J. Dermatol. 2009, 160, 80–91. [Google Scholar] [PubMed]
- Dias, M.; Hadgraft, J.; Lane, M.E. Influence of membrane-solvent-solute interactions on solute permeation in skin. Int. J. Pharm. 2007, 340, 65–70. [Google Scholar]
- Oliveira, G.; Hadgraft, J.; Lane, M.E. The influence of volatile solvents on transport across model membranes and human skin. Int. J. Pharm. 2012, 435, 38–49. [Google Scholar] [PubMed]
- Lane, M.E.; Hadgraft, J.; Oliveira, G.; Vieira, R.; Mohammed, D.; Hirata, K. Rational formulation design. Int. J. Cosmet. Sci. 2012, 34, 496–501. [Google Scholar]
- Bielfeldt, S.; Bonnier, F.; Byrne, H.J.; Chourpa, I.; Dancik, Y.; Lane, M.E.; Lunter, D.J.; Munnier, E.; Puppels, G.; Tfayli, A.; et al. Monitoring dermal penetration and permeation kinetics of topical products; the role of Raman microspectroscopy. TrAC Trends Anal. Chem. 2022, 156, 116709. [Google Scholar]
- Franzen, L.; Selzer, D.; Fluhr, J.W.; Schaefer, U.F.; Windbergs, M. Towards drug quantification in human skin with confocal Raman microscopy. Eur. J. Pharm. Biopharm. 2013, 84, 437–444. [Google Scholar]
- Badruddoza, A.Z.M.; Yeoh, T.; Shah, J.C.; Walsh, T. Assessing and Predicting Physical Stability of Emulsion-Based Topical Semisolid Products: A Review. J. Pharm. Sci. 2023, 112, 1772–1793. [Google Scholar]
- Surber, C.; Schmid-Grendelmeier, P. General principles of topical therapy of the skin. In EAACI Global Atlas of Allergy; Akdis, C.A., Agache, I., Jutel, M., Hellings, P., Schmidt-Weber, P., Schmid-Grendelmeier, P., Hoffmann-Sommergruber, K., Muraro, A., Bindslev-Jensen, C., Torres, M., et al., Eds.; European Academy of Allergy and Clinical Immunology: Zurich, Switzerland, 2019. [Google Scholar]
- Goyal, R.; Macri, L.K.; Kaplan, H.M.; Kohn, J. Nanoparticles and nanofibers for topical drug delivery. J. Control. Release 2016, 240, 77–92. [Google Scholar]
- Roberts, M.S.; Mohammed, Y.; Pastore, M.N.; Namjoshi, S.; Yousef, S.; Alinaghi, A.; Haridass, I.N.; Abd, E.; Leite-Silva, V.R.; Benson, H.; et al. Topical and cutaneous delivery using nanosystems. J. Control Release 2017, 247, 86–105. [Google Scholar]
- Busch, L.; Keziban, Y.; Dähne, L.; Keck, C.M.; Meinke, M.C.; Lademann, J.; Patzelt, A. The impact of skin massage frequency on the intrafollicular transport of silica nanoparticles: Validation of the ratchet effect on an ex vivo porcine skin model. Eur. J. Pharm. Biopharm. 2021, 158, 266–272. [Google Scholar]
- Patzelt, A.; Richter, H.; Knorr, F.; Schäfer, U.; Lehr, C.-M.; Dähne, L.; Sterry, W.; Lademann, J. Selective follicular targeting by modification of the particle sizes. J. Control. Release 2011, 150, 45–48. [Google Scholar]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), ICH Quality Guidelines. Available online: https://www.ich.org/page/quality-guidelines (accessed on 9 June 2023).
- European Medicines Agency. ICH Topic Q 6 A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances; European Medicines Agency: Amsterdam, The Netherlands, 2000. [Google Scholar]
- World Health Organization (WHO). Stability conditions for WHO Member States by Region; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- European Parliament. Directive 2001/83/EC on the Community Code Relating to Medicinal Products for Human Use; European Parliament: Strasbourg, France, 2001. [Google Scholar]
- De Winter, S.; Vanbrabant, P.; Vi, N.T.T.; Deng, X.; Spriet, I.; van Schepdael, A.; Gillet, J.-B. Impact of temperature exposure on stability of drugs in a real-world out-of-hospital setting. Ann. Emerg. Med. 2013, 62, 380–387.e1. [Google Scholar]
- Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 2 May 2023).
- Yoshida, H.; Tamura, S.; Toyoda, T.; Kado, K.; Ohnishi, N.; Ibuki, R. In vitro release of Tacrolimus from Tacrolimus ointment and its speculated mechanism. Int. J. Pharm. 2004, 270, 55–64. [Google Scholar] [CrossRef]
- Surber, C.; Robertis, J.; Reinau, D. Topical corticosteroid or emollient product: Which to apply first? J. Eur. Acad. Dermatol. Venereol. JEADV 2022, 37, e646–e647. [Google Scholar] [CrossRef]
- Daniels, R.; Knie, U. Galenics of dermal products—Vehicles, properties and drug release. J. Der Dtsch. Dermatol. Ges. 2007, 5, 367–383. [Google Scholar]
- EMA/356530/2021; Klisyri, Summary of Product Characteristics. European Medicines Agency: Amsterdam, The Netherlands, 2022.
- Lane, M.E. Skin penetration enhancers. Int. J. Pharm. 2013, 447, 12–21. [Google Scholar]
- Thompson, J.E. (Ed.) A Practical Guide to Contemporary Pharmacy Practice; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009. [Google Scholar]
- Großmann, A. Influence of Manufacturing Process Parameters on the Physical Properties of an Oil-in-Water Cream. Doctor’s Dissertation, Universität Tübingen, Tübingen, Germany, 2007. [Google Scholar]
- Ballmann, C. Entwicklung und Charakterisierung halbfester Zubereitungen auf der Basis von Triglyceriden. Doctor’s Dissertation, Christian-Albrechts-Universität zu Kiel, Kiel, Germany, 2006. [Google Scholar]
- Puschmann, J.; Herbig, M.E.; Müller-Goymann, C.C. Influence of emulsifier concentration on partition behavior and chemical stability of betamethasone dipropionate in emulsion gels. Int. J. Pharm. 2019, 562, 105–112. [Google Scholar]
- Evers, D.-H.; Fielhauer, S.; Gorissen, S.; Hahn, S.; Herbig, M.; Köllmer, M. Pharmaceutical Composition Comprising Tacrolimus. WO2019233722, 12 December 2019. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Choy, Y.B.; Prausnitz, M.R. The rule of five for non-oral routes of drug delivery: Ophthalmic, inhalation and transdermal. Pharm. Res. 2011, 28, 943–948. [Google Scholar]
- RGuy, H.; Potts, R.O. Structure-permeability relationships in percutaneous penetration. J. Pharm. Sci. 1992, 81, 603–604. [Google Scholar]
- Abraham, M.H.; Martins, F. Human skin permeation and partition: General linear free-energy relationship analyses. J. Pharm. Sci. 2004, 93, 1508–1523. [Google Scholar] [PubMed]
- Potts, R.O.; Guy, R.H. Predicting skin permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [PubMed]
- Faller, B.; Ertl, P. Computational approaches to determine drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 533–545. [Google Scholar] [PubMed]
- Schuetz, Y.B.; Carrupt, P.-A.; Naik, A.; Guy, R.H.; Kalia, Y.N. Structure-permeation relationships for the non-invasive transdermal delivery of cationic peptides by iontophoresis. Eur. J. Pharm. Sci. 2006, 29, 53–59. [Google Scholar]
- Santos, L.L.; Wu, E.L.; Grinias, K.M.; Koetting, M.C.; Jain, P. Developability profile framework for lead candidate selection in topical dermatology. Int. J. Pharm. 2021, 604, 120750. [Google Scholar]
- Davis, A.F.; Hadgraft, J. Effect of supersaturation on membrane transport: 1. Hydrocortisone acetate. Int. J. Pharm. 1991, 76, 1–8. [Google Scholar]
- Wiechers, J.W.; Kelly, C.L.; Blease, T.G.; Dederen, J.C. Formulating for efficacy. Int. J. Cosmet. Sci. 2004, 26, 173–182. [Google Scholar]
- Abbott, S. An integrated approach to optimizing skin delivery of cosmetic and pharmaceutical actives. Int. J. Cosmet. Sci. 2012, 34, 217–222. [Google Scholar] [CrossRef]
- Yamanaka, M.; Yokota, S.; Iwao, Y.; Noguchi, S.; Itai, S. Development and evaluation of a tacrolimus cream formulation using a binary solvent system. Int. J. Pharm. 2014, 464, 19–26. [Google Scholar]
- Reid, M.L.; Jones, S.A.; Brown, M.B. Transient drug supersaturation kinetics of beclomethasone dipropionate in rapidly drying films. Int. J. Pharm. 2009, 371, 114–119. [Google Scholar]
- Moser, K.; Kriwet, K.; Froehlich, C.; Kalia, Y.N.; Guy, R.H. Supersaturation: Enhancement of skin penetration and permeation of a lipophilic drug. Pharm. Res. 2001, 18, 1006–1011. [Google Scholar] [CrossRef]
- Frederiksen, K.; Guy, R.H.; Petersson, K. Formulation considerations in the design of topical, polymeric film-forming systems for sustained drug delivery to the skin. Eur. J. Pharm. Biopharm. 2015, 91, 9–15. [Google Scholar]
- Kapoor, K.; Gräfe, N.; Herbig, M.E. Topical Film-Forming Solid Solutions for Enhanced Dermal Delivery of the Retinoid Tazarotene. J. Pharm. Sci. 2022, 111, 2779–2787. [Google Scholar]
- Herrmann, S.; Daniels, R.; Lunter, D. Methods for the determination of the substantivity of topical formulations. Pharm. Dev. Technol. 2017, 22, 487–491. [Google Scholar]
- Puschmann, J.; Herbig, M.E.; Müller-Goymann, C.C. Correlation between free aqueous preservative concentration in emulsion gels measured by equilibrium dialysis and antimicrobial efficacy. Eur. J. Pharm. Biopharm. 2018, 131, 152–161. [Google Scholar]
- Peralta, M.F.; Guzmán, M.L.; Pérez, A.P.; Apezteguia, G.A.; Fórmica, M.L.; Romero, E.L.; Olivera, M.E.; Carrer, D.C. Liposomes can both enhance or reduce drugs penetration through the skin. Sci. Rep. 2018, 8, 13253. [Google Scholar]
- Bouwstra, J.; Honeywell-Nguyen, P. Skin structure and mode of action of vesicles. Adv. Drug Deliv. Rev. 2002, 54, S41–S55. [Google Scholar]
- Honeywell-Nguyen, P.L.; Bouwstra, J.A. The in vitro transport of pergolide from surfactant-based elastic vesicles through human skin: A suggested mechanism of action. J. Control. Release 2003, 86, 145–156. [Google Scholar]
- European Medicines Agency. Q8 (R2) Step 5 Pharmaceutical Development; European Medicines Agency: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Vishwasrao, S.; Singh, S. Current Perspective on Opportunities and Adoption Challenges of QbD Implementation in Pharmaceutical Product Development. Pharm. Process Dev. 2016, 2016, 1–8. [Google Scholar]
- Namjoshi, S.; Dabbaghi, M.; Roberts, M.S.; Grice, J.E.; Mohammed, Y. Quality by Design: Development of the Quality Target Product Profile (QTPP) for Semisolid Topical Products. Pharmaceutics 2020, 12, 287. [Google Scholar] [PubMed]
- Simões, A.; Veiga, F.; Vitorino, C.; Figueiras, A. A Tutorial for Developing a Topical Cream Formulation Based on the Quality by Design Approach. J. Pharm. Sci. 2018, 107, 2653–2662. [Google Scholar] [PubMed]
- Evers, D.-H.; Schultz-Fademrecht, T.; Garidel, P.; Buske, J. Development and validation of a selective marker-based quantification of polysorbate 20 in biopharmaceutical formulations using UPLC QDa detection. J. Chromatogr. B 2020, 1157, 122287. [Google Scholar]
- Council of Europe (Ed). European Pharmacopoeia, 11th ed.; Council of Europe: Strasbourg, France, 2023; pp. 3207–3210. [Google Scholar]
- Langley, N.; Michniak-Kohn, B.; Osborne, D.W. (Eds.) The Role of Microstructure in Topical Drug Product Development; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Rawat, A.; Gupta, S.S.; Kalluri, H.; Lowenborg, M.; Bhatia, K.; Warner, K. Rheological Characterization in the Development of Topical Drug Products. In The Role of Microstructure in Topical Drug Product Development; Langley, N., Michniak-Kohn, B., Osborne, D.W., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 3–45. [Google Scholar]
- Brummer, R. Rheology Essentials of Cosmetic and Food Emulsions; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2016. [Google Scholar]
- CHMP/QWP/708282/2018; Draft Guideline on Quality and Equivalence of Topical Products. European Medicines Agency: Amsterdam, The Netherlands, 2018.
- Dong, L.; Liu, C.; Cun, D.; Fang, L. The effect of rheological behavior and microstructure of the emulgels on the release and permeation profiles of Terpinen-4-ol. Eur. J. Pharm. Sci. 2015, 78, 140–150. [Google Scholar]
- Binder, L.; Mazál, J.; Petz, R.; Klang, V.; Valenta, C. The role of viscosity on skin penetration from cellulose ether-based hydrogels. Ski. Res. Technol. 2019, 25, 725–734. [Google Scholar] [CrossRef]
- Calixto, L.S.; Infante, V.H.P.; Campos, P.M.B.G.M. Design and Characterization of Topical Formulations: Correlations Between Instrumental and Sensorial Measurements. AAPS PharmSciTech 2018, 19, 1512–1519. [Google Scholar]
- Moravkova, T.; Filip, P. Relation between sensory analysis and rheology of body lotions. Int. J. Cosmet. Sci. 2016, 38, 558–566. [Google Scholar]
- Sailer, M.M.; Köllmer, M.; Masson, B.; Fais, F.; Hohenfeld, I.P.; Herbig, M.E.; Koitschev, A.K.; Becker, S. Nasal residence time and rheological properties of a new bentonite-based thixotropic gel emulsion nasal spray—AM-301. Drug Dev. Ind. Pharm. 2023, 49, 103–114. [Google Scholar] [CrossRef]
- Bakshi, M.; Singh, S. Development of validated stability-indicating assay methods—Critical review. J. Pharm. Biomed. Anal. 2002, 28, 1011–1040. [Google Scholar]
- Blessy, M.; Patel, R.D.; Prajapati, P.N.; Agrawal, Y.K. Development of forced degradation and stability indicating studies of drugs—A review. J. Pharm. Anal. 2014, 4, 159–165. [Google Scholar]
- European Medicines Agency. CHMP/ICH, Q 3 B (R2) Impurities in New Drug Products; European Medicines Agency: Amsterdam, The Netherlands, 2006. [Google Scholar]
- European Medicines Agency. CHMP/ICH, Q 1 A (R2) Stability Testing of new Drug Substances and Products; European Medicines Agency: Amsterdam, The Netherlands, 2003. [Google Scholar]
- USP-NF. 1220 Analytical Procedure Life Cycle; USP-NF: North Bethesda, ML, USA, 2022. [Google Scholar]
- Franz, T.J. Percutaneous absorption on the relevance of in vitro data. J. Investig. Dermatol. 1975, 64, 190–195. [Google Scholar]
- Lunter, D.; Daniels, R. In Vitro skin permeation and penetration of nonivamide from novel film-forming emulsions. Ski. Pharmacol. Physiol. 2013, 26, 139–146. [Google Scholar]
- Carrer, V.; Alonso, C.; Oliver, M.A.; Coderch, L. In Vitro penetration through the skin layers of topically applied glucocorticoids. Drug Test. Anal. 2018, 10, 1528–1535. [Google Scholar]
- Raney, S.G.; Ghosh, P.; Ramezanli, T.; Lehman, P.A.; Franz, T.J. Cutaneous Pharmacokinetic Approaches to Compare Bioavailability and/or Bioequivalence for Topical Drug Products. Dermatol. Clin. 2022, 40, 319–332. [Google Scholar]
- Miranda, M.; Veloso, C.; Brown, M.; Pais, A.A.C.C.; Cardoso, C.; Vitorino, C. Topical bioequivalence: Experimental and regulatory considerations following formulation complexity. Int. J. Pharm. 2022, 620, 121705. [Google Scholar]
- Herbig, M.E.; Houdek, P.; Gorissen, S.; Zorn-Kruppa, M.; Wladykowski, E.; Volksdorf, T.; Grzybowski, S.; Kolios, G.; Willers, C.; Mallwitz, H.; et al. A custom tailored model to investigate skin penetration in porcine skin and its comparison with human skin. Eur. J. Pharm. Biopharm. 2015, 95, 99–109. [Google Scholar]
- Quartier, J.; Capony, N.; Lapteva, M.; Kalia, Y.N. Cutaneous Biodistribution: A High-Resolution Methodology to Assess Bioequivalence in Topical Skin Delivery. Pharmaceutics 2019, 11, 484. [Google Scholar]
- Holmgaard, R.; Benfeldt, E.; Nielsen, J.B.; Gatschelhofer, C.; Sorensen, J.A.; Höfferer, C.; Bodenlenz, M.; Pieber, T.R.; Sinner, F. Comparison of open-flow microperfusion and microdialysis methodologies when sampling topically applied fentanyl and benzoic acid in human dermis ex vivo. Pharm. Res. 2012, 29, 1808–1820. [Google Scholar]
- Mohammed, D.; Matts, P.J.; Hadgraft, J.; Lane, M.E. Depth profiling of stratum corneum biophysical and molecular properties. Br. J. Dermatol. 2011, 164, 957–965. [Google Scholar]
- N’Dri-Stempfer, B.; Navidi, W.C.; Guy, R.H.; Bunge, A.L. Improved bioequivalence assessment of topical dermatological drug products using dermatopharmacokinetics. Pharm. Res. 2009, 26, 316–328. [Google Scholar]
- Pensado, A.; Chiu, W.S.; Cordery, S.F.; Rantou, E.; Bunge, A.L.; Delgado-Charro, M.B.; Guy, R.H. Stratum Corneum Sampling to Assess Bioequivalence between Topical Acyclovir Products. Pharm. Res. 2019, 36, 180. [Google Scholar] [PubMed]
- Liu, Y.; Lunter, D.J. Confocal Raman spectroscopy at different laser wavelengths in analyzing stratum corneum and skin penetration properties of mixed PEGylated emulsifier systems. Int. J. Pharm. 2022, 616, 121561. [Google Scholar] [PubMed]
- Lunter, D.; Daniels, R. Confocal Raman microscopic investigation of the effectiveness of penetration enhancers for procaine delivery to the skin. J. Biomed. Opt. 2014, 19, 126015. [Google Scholar] [PubMed]
- Iliopoulos, F.; Tang, C.F.; Li, Z.; Rahma, A.; Lane, M.E. Confocal Raman Spectroscopy for Assessing Bioequivalence of Topical Formulations. Pharmaceutics 2023, 15, 1075. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trade Name | Eucrisa® | Aklief® | Winlevi® | Klisyri® | Opzelura® | VTAMA® | ZORYVE® | |
|---|---|---|---|---|---|---|---|---|
| API and Concentration | Crisaborole 2% | Trifarotene 0.005% | Clascoterone 1% | Tirbanibulin 1% | Ruxolitinib 1.5% | Tapinarof 1% | Roflumilast 0.3% | |
| Indication | Atopic Dermatitis | Acne Vulgaris | Acne Vulgaris | Actinic Keratosis | Atopic Dermatitis | Plaque Psoriasis | Plaque Psoriasis | |
| Formulation Type | Ointment | Cream | Cream | Ointment | Cream | Cream | Cream | |
| Year of US Approval | 2016 | 2019 | 2020 | 2020 | 2021 | 2022 | 2022 | |
| Excipient | Properties/Typical Function | |||||||
| White petrolatum | Ointment base/oil phase | x | x | x | ||||
| Paraffin | Oil phase | x | ||||||
| Light mineral oil | Oil phase | x | ||||||
| Mineral oil | Oil phase | x | ||||||
| Medium-chain triglycerides | Oil phase | x | x | x | ||||
| Isopropyl palmitate | Oil phase | x | ||||||
| Cyclomethicone | Oil phase | x | ||||||
| Dimethicone 350 | Oil phase | x | ||||||
| Polyethylene glycol 200 | Solvent | x | ||||||
| Hexylene glycol | Solvent | x | ||||||
| Propylene glycol | Solvent/penetration enhancer | x | x | x | x | x | x | |
| Diethylene glycol monoethyl ether | Solvent/penetration enhancer | x | x | |||||
| Purified water | Hydrophilic solvent | x | x | x | x | x | ||
| Ethanol | Hydrophilic solvent | x | ||||||
| Emulsifying wax | Emulsifier/consistency agent | x | ||||||
| Glyceryl stearate SE | Emulsifier/consistency agent | x | ||||||
| Mono- and di-glycerides | Consistency agent/co-emulsifier | x | x | x | ||||
| Cetyl alcohol | Consistency agent/co-emulsifier | x | x | |||||
| Cetostearyl alcohol | Consistency agent/co-emulsifier | x | ||||||
| Stearyl alcohol | Consistency agent/co-emulsifier | x | ||||||
| Polysorbate 20 | Emulsifier/solubilizer | x | ||||||
| Polysorbate 80 | Emulsifier/solubilizer | x | x | |||||
| Polyoxyl 2 stearyl ether | Emulsifier | x | ||||||
| Polyoxyl 20 stearyl ether | Emulsifier | x | ||||||
| Ceteareth-10 phosphate | Emulsifier | x | ||||||
| Cetearyl phosphate | Emulsifier | x | ||||||
| Xanthan gum | Thickener | x | ||||||
| Copolymer acrylamide and acryloyldimethyl taurate * | Thickening and emulsifying polymer | x | ||||||
| Allantoin | Humectant | x | ||||||
| Phenoxyethanol | Preservative | x | x | |||||
| Methylparaben | Preservative | x | x | |||||
| Propylparaben | Preservative | x | x | |||||
| Benzoic acid | Preservative | x | ||||||
| Butylated hydroxytoluene | Antioxidant | x | x | |||||
| Vitamine E | Antioxidant | x | ||||||
| Edetate calcium | Chelating agent | x | ||||||
| Edetate disodium | Chelating agent | x | x | |||||
| Citric acid monohydrate | Buffer | x | x | |||||
| Sodium citrate dihydrate | Buffer | x | ||||||
| Sodium hydroxide | pH adjustment | x | ||||||
| Solvent | Solubility of Tacrolimus [mg/mL] |
|---|---|
| Water and aqueous solutions | |
| Water | <0.001 |
| Co-Solvents | |
| Triethyl citrate | 105.2 |
| Oil phases | |
| Medium chain triglycerides (MCT) | 10.8 |
| Diisopropyl adipate (DIPA) | 63.3 |
| Isopropyl myristate | 4.1 |
| Liquid paraffin | 0.007 |
| Composed oil phases | |
| DIPA:MCT (50:50) | 26.4 |
| DIPA:paraffin:MCT (25:25:50) | 10.1 |
| Parameter | IVRT | IVPT |
|---|---|---|
| Investigated process | Release | Permeation |
| Membrane used | Synthetic filter membrane | (human) skin |
| Donor chamber exposure | Occluded | Often unoccluded |
| Dosing | Infinite dose | Often (semi)finite dose |
| Readout parameters | Flux profile (Jmax, etc.) | Release rate (slope) |
| Receptor cell media | Non-physiological media acceptable | Physiological media preferred |
| Typcial detection range | µg/mL range | ng/mL range |
| Contact of product with acceptor | Product-media interface | Product stays “dry“ |
| Variability in membrane | Relatively consistent in quality | Donor variability |
| in vitro/in vivo correlation | Not designed to correlate with in vivo | IVIV correlation expected |
| Overall purpose | Assessment of quality | Assessment of performance/safety |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbig, M.E.; Evers, D.-H.; Gorissen, S.; Köllmer, M. Rational Design of Topical Semi-Solid Dosage Forms-How Far Are We? Pharmaceutics 2023, 15, 1822. https://doi.org/10.3390/pharmaceutics15071822
Herbig ME, Evers D-H, Gorissen S, Köllmer M. Rational Design of Topical Semi-Solid Dosage Forms-How Far Are We? Pharmaceutics. 2023; 15(7):1822. https://doi.org/10.3390/pharmaceutics15071822
Chicago/Turabian StyleHerbig, Michael E., Dirk-Heinrich Evers, Sascha Gorissen, and Melanie Köllmer. 2023. "Rational Design of Topical Semi-Solid Dosage Forms-How Far Are We?" Pharmaceutics 15, no. 7: 1822. https://doi.org/10.3390/pharmaceutics15071822
APA StyleHerbig, M. E., Evers, D.-H., Gorissen, S., & Köllmer, M. (2023). Rational Design of Topical Semi-Solid Dosage Forms-How Far Are We? Pharmaceutics, 15(7), 1822. https://doi.org/10.3390/pharmaceutics15071822