
Clinical Advances and Perspectives in Targeted Radionuclide Therapy



Abstract
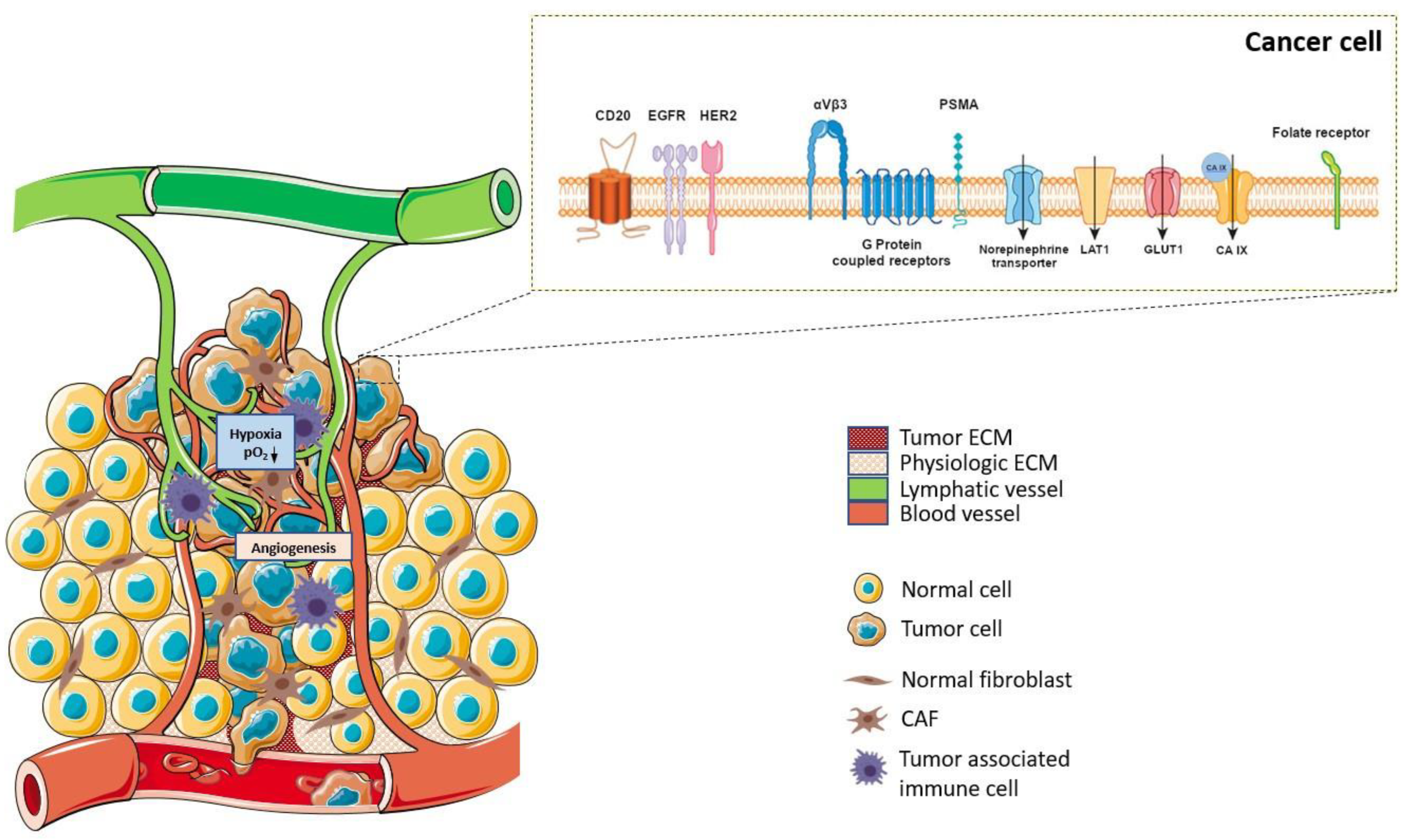
1. Introduction
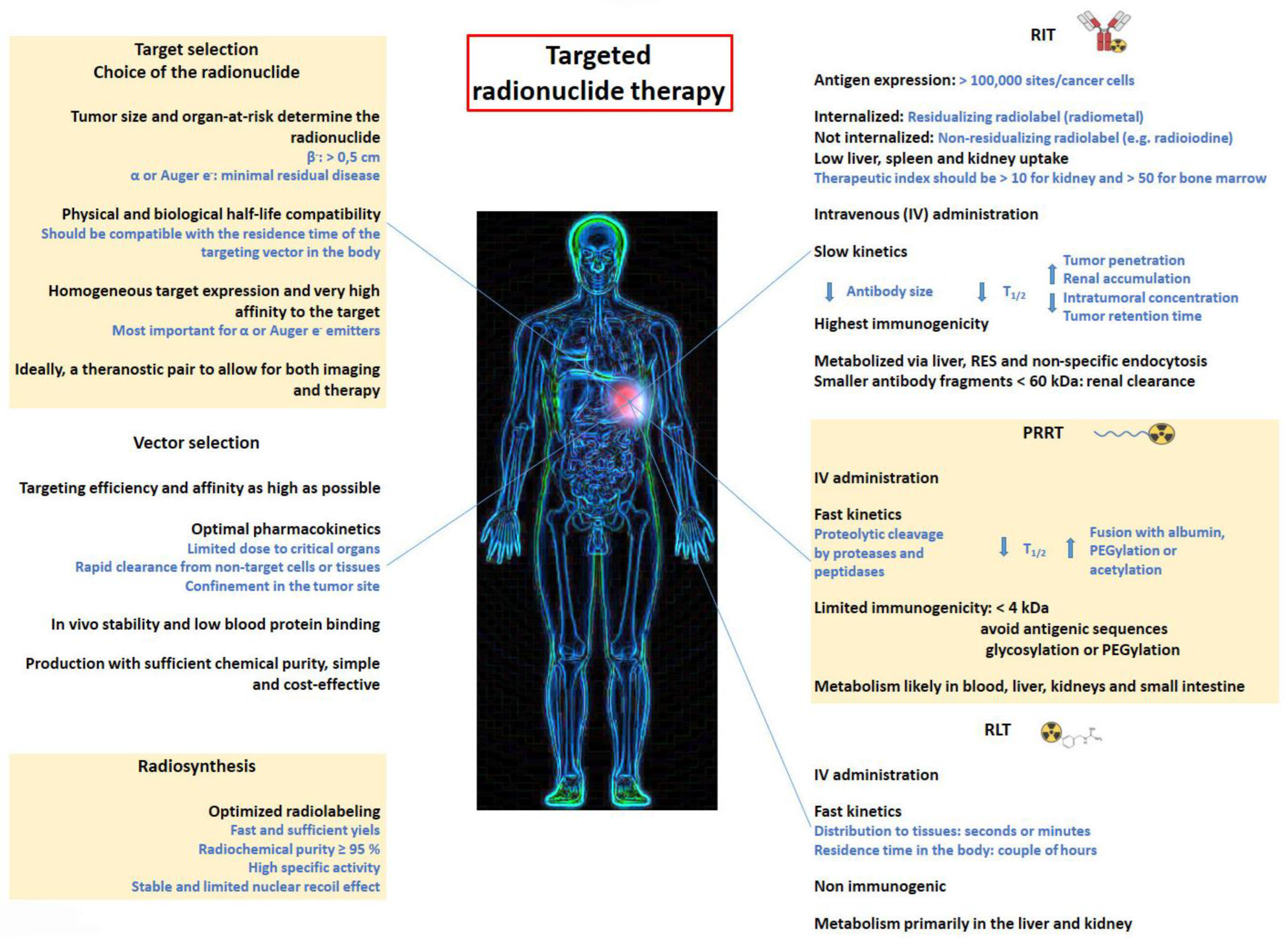
2. Radioimmunotherapy (RIT)
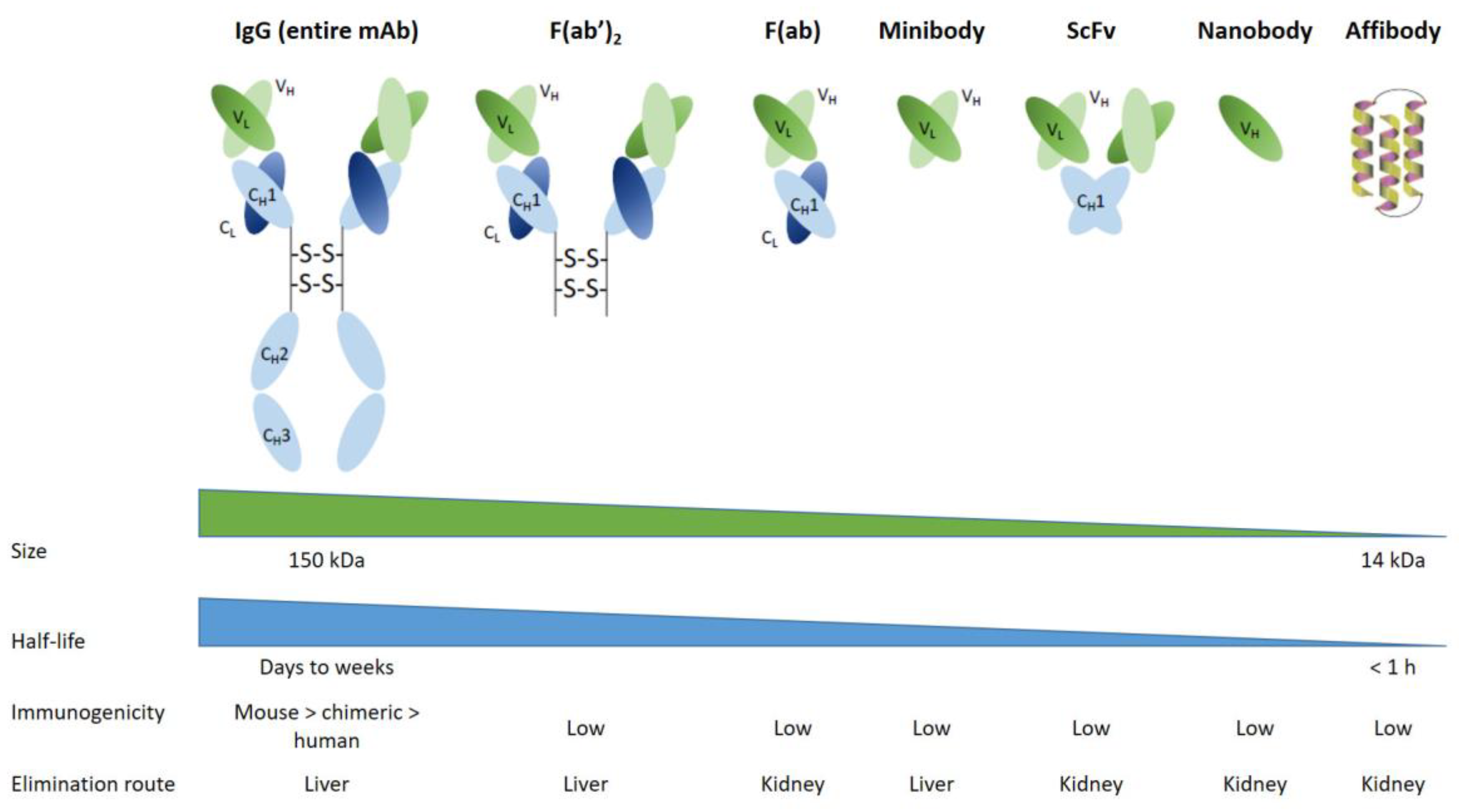
2.1. Antibodies and Derivatives
2.2. Pretargeting Approach
2.3. Dose Fractionation Approach
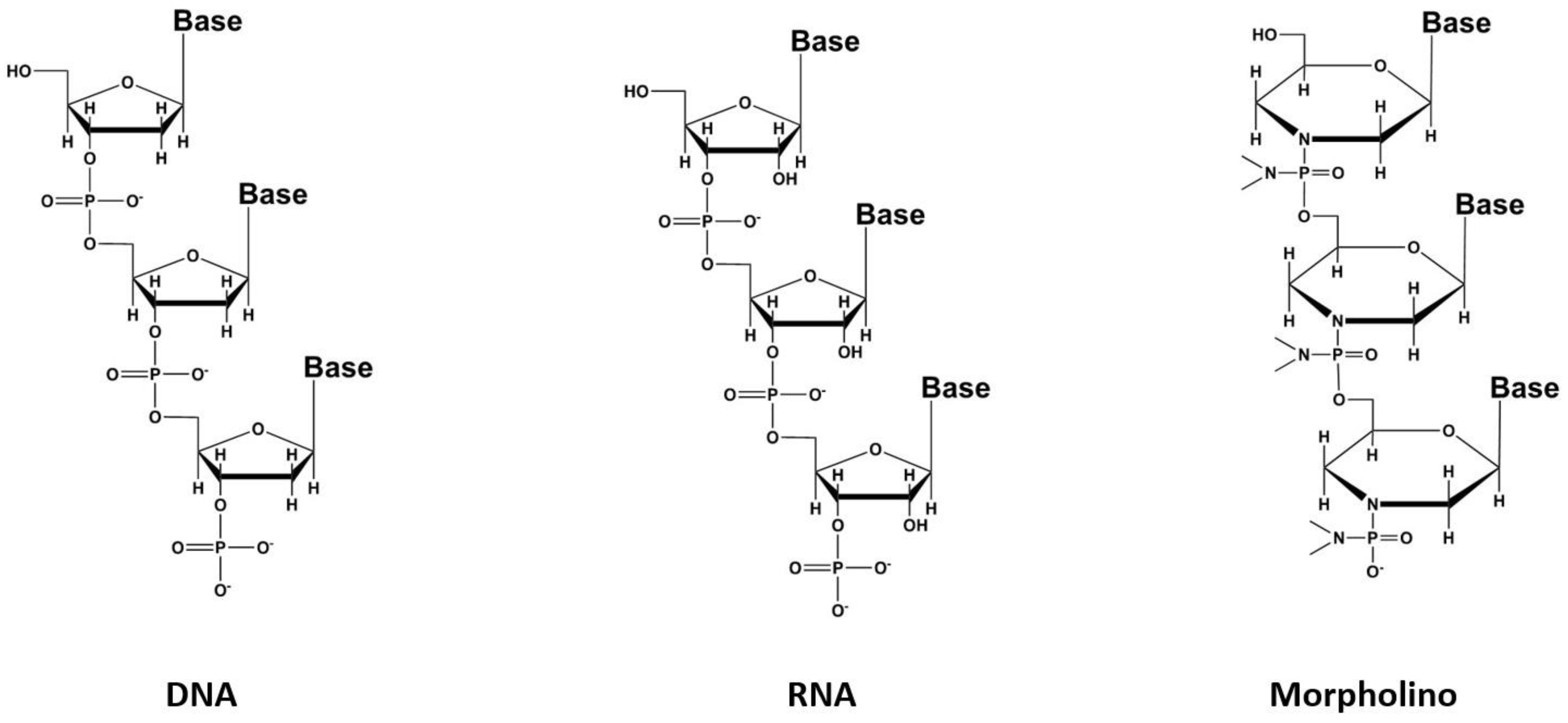
3. Oligonucleotides
4. Peptide Receptor Radionuclide Therapy (PRRT)
4.1. The G Protein-Coupled Receptors Family
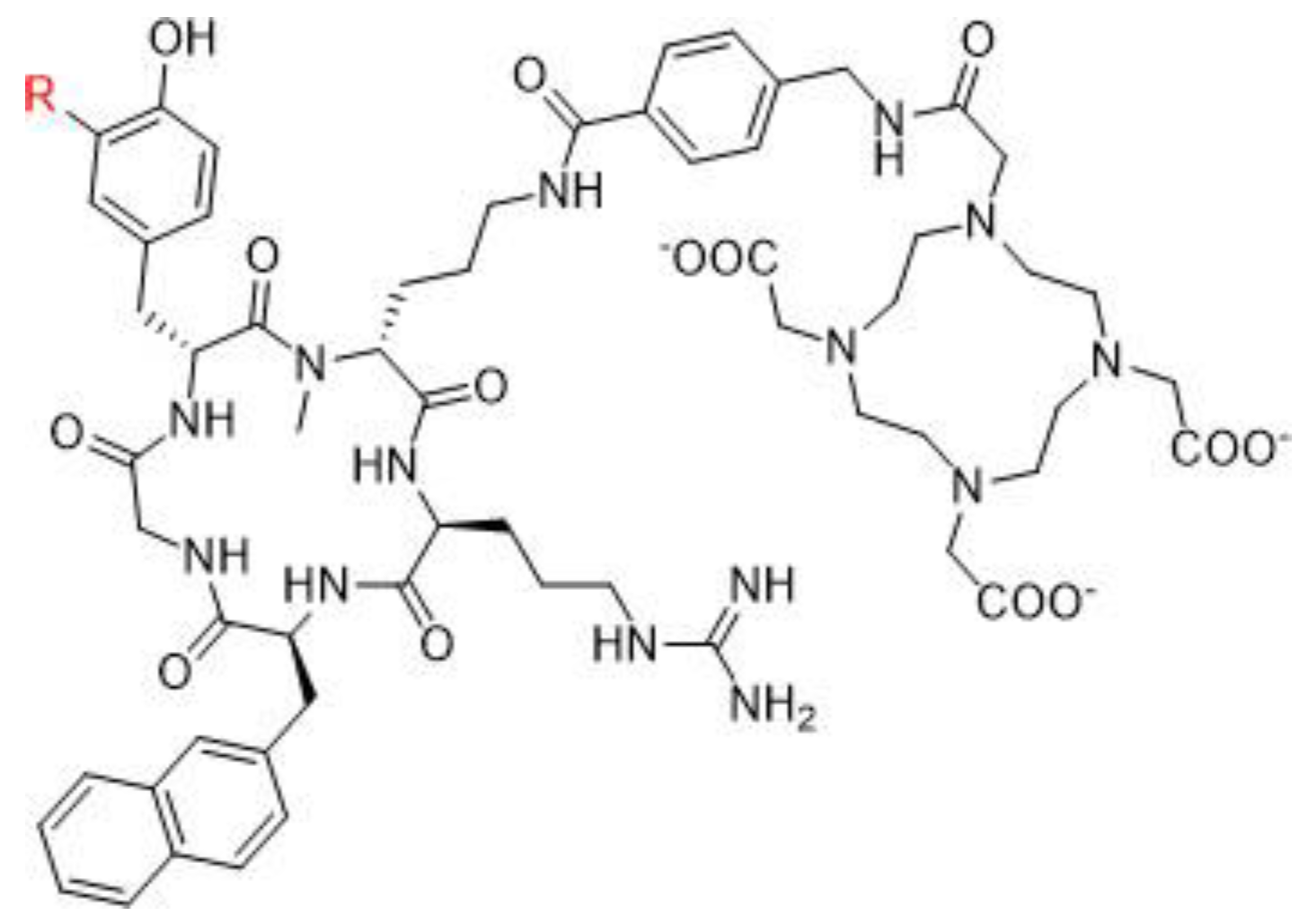
4.1.1. Somatostatin Analogs
4.1.2. Bombesin Analogs
4.1.3. Substance P
4.1.4. Other Analogs
4.2. C-X-C Chemokine Receptor Type 4 (CXCR-4)
4.3. Other Peptide Derivatives
5. Radioligand Therapy (RLT)
5.1. Bone-Seeking Agents
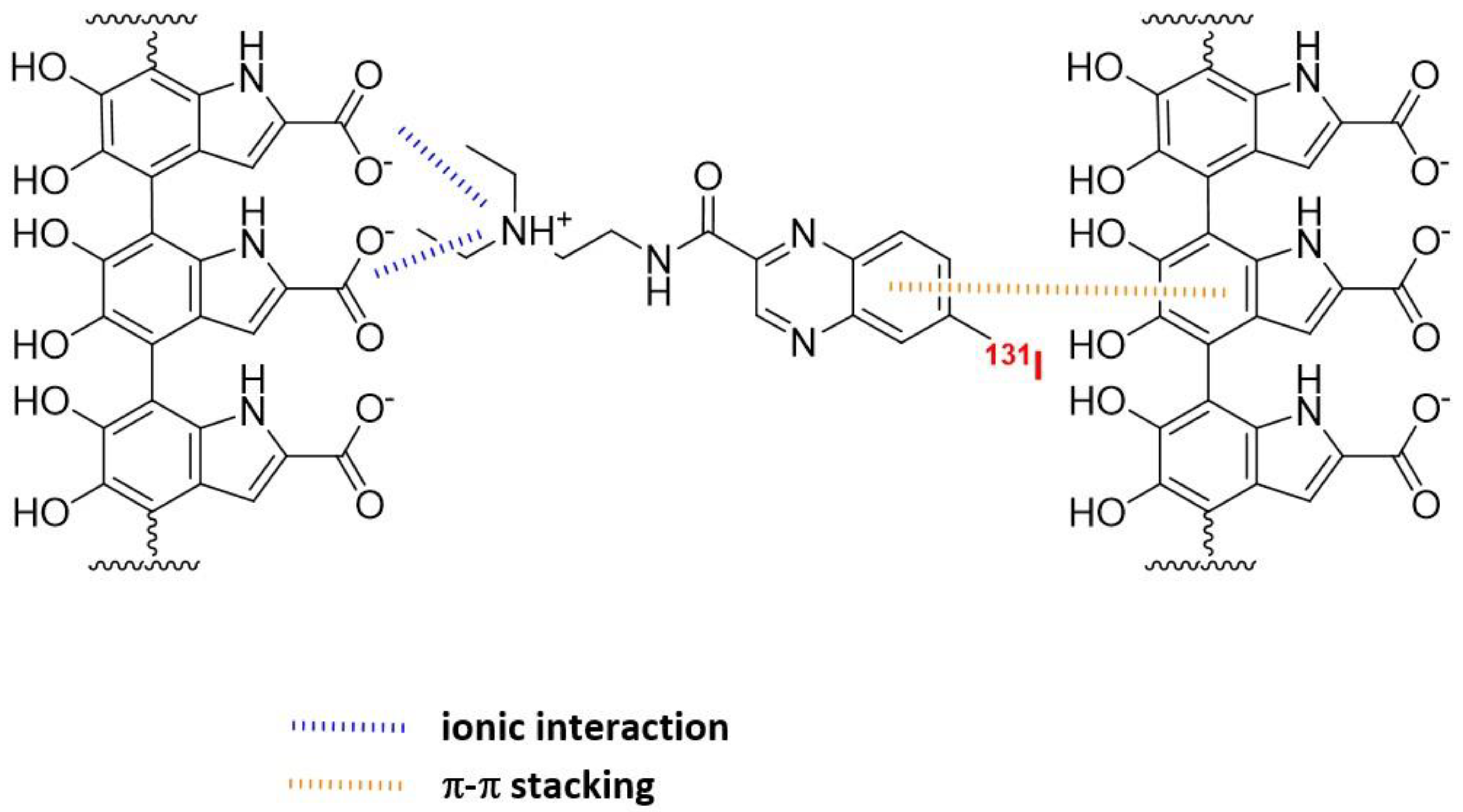
5.2. [131I]mIBG
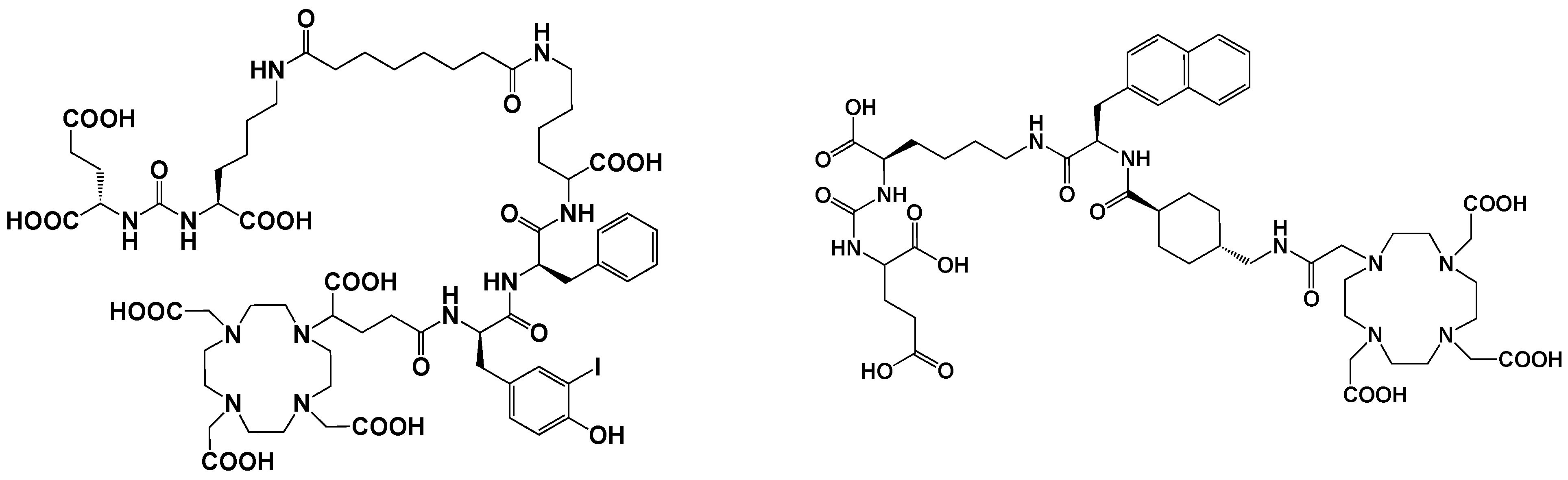
5.3. Prostate-Specific Membrane Antigen (PSMA) Inhibitors
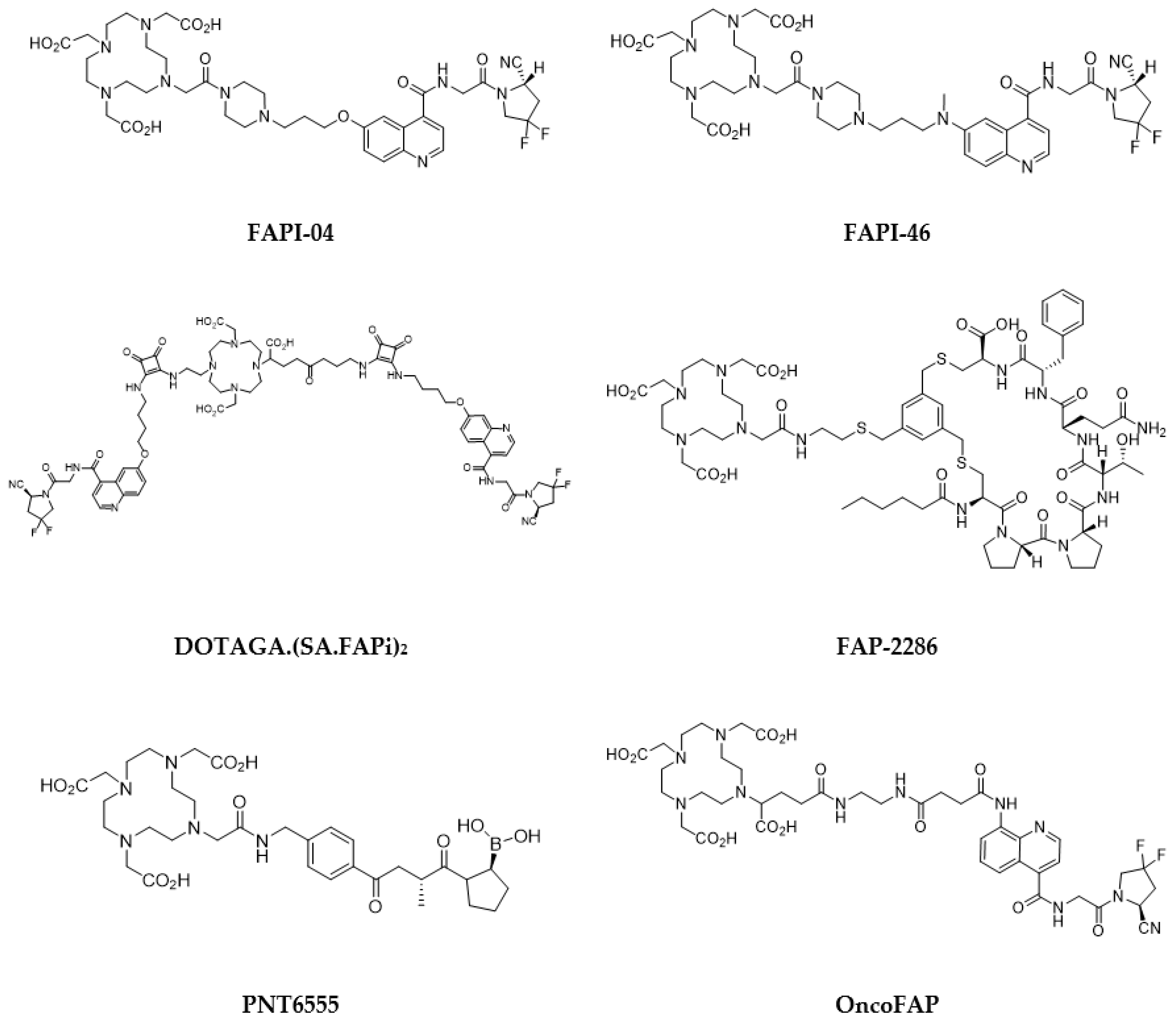
5.4. Fibroblast-Activation Protein (FAP) Inhibitors
5.5. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors
5.6. Carbonic Anhydrase IX (CA IX) Inhibitors
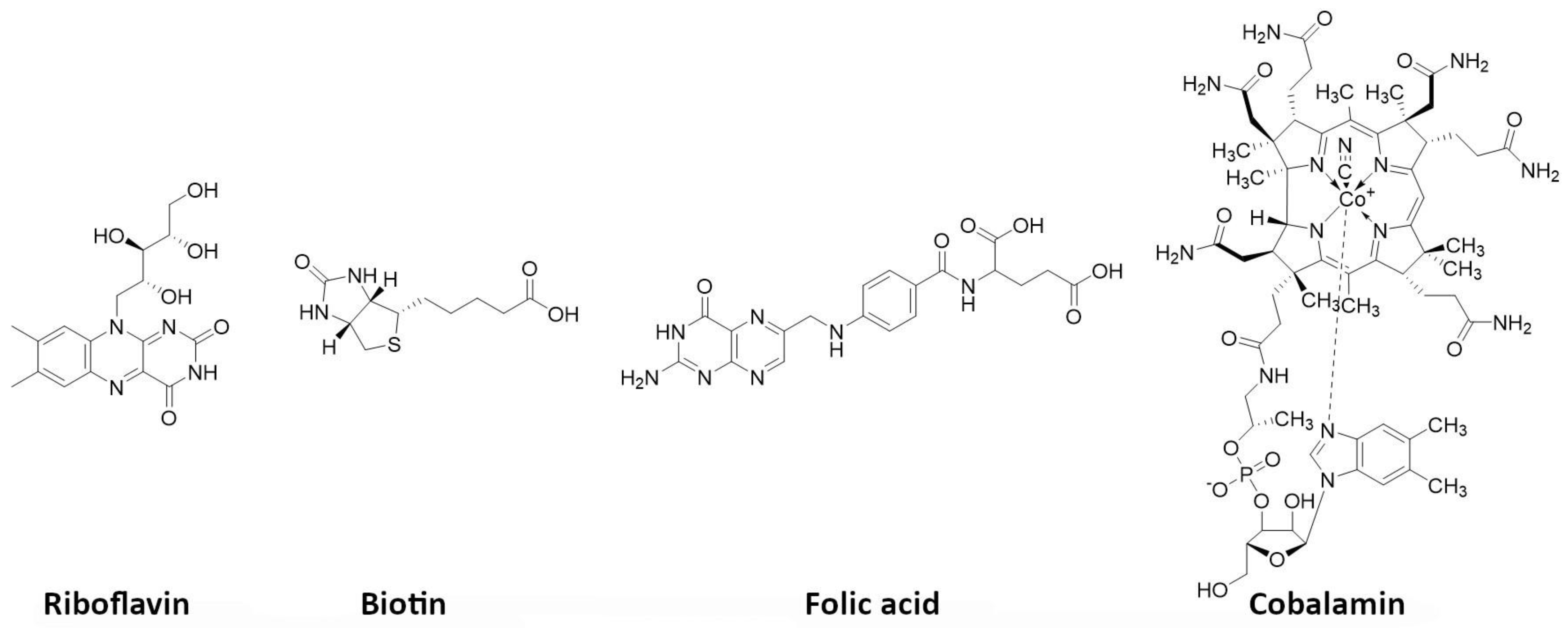
5.7. Vitamins
5.8. Phospholipid Ether Analogues
5.9. Melanin Targeting Agents
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hertz, B.; Watabe, T.; Baum, R.P. Celebrating 80 years anniversary of radioiodine for use in thyroid cancer and perspectives for theranostics. Ann. Nucl. Med. 2022, 36, 1007–1009. [Google Scholar] [CrossRef] [PubMed]
- Volkert, W.A.; Hoffman, T.J. Therapeutic radiopharmaceuticals. Chem. Rev. 1999, 99, 2269–2292. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, M. Radionuclide therapy beyond radioiodine. Wien. Med. Wochenschr. 2012, 162, 430–439. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef] [PubMed]
- St James, S.; Bednarz, B.; Benedict, S.; Buchsbaum, J.C.; Dewaraja, Y.; Frey, E.; Hobbs, R.; Grudzinski, J.; Roncali, E.; Sgouros, G.; et al. Current Status of Radiopharmaceutical Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 891–901. [Google Scholar] [CrossRef]
- Pouget, J.P.; Santoro, L.; Raymond, L.; Chouin, N.; Bardiès, M.; Bascoul-Mollevi, C.; Huguet, H.; Azria, D.; Kotzki, P.O.; Pèlegrin, M.; et al. Cell membrane is a more sensitive target than cytoplasm to dense ionization produced by auger electrons. Radiat. Res. 2008, 170, 192–200. [Google Scholar] [CrossRef]
- Fernandes, C.; Palma, E.; Silva, F.; Belchior, A.; Pinto, C.I.G.; Guerreiro, J.F.; Botelho, H.M.; Mendes, F.; Raposinho, P.; Paulo, A. Searching for a Paradigm Shift in Auger-Electron Cancer Therapy with Tumor-Specific Radiopeptides Targeting the Mitochondria and/or the Cell Nucleus. Int. J. Mol. Sci. 2022, 23, 7238. [Google Scholar] [CrossRef]
- Pirovano, G.; Wilson, T.C.; Reiner, T. Auger: The future of precision medicine. Nucl. Med. Biol. 2021, 96–97, 50–53. [Google Scholar] [CrossRef]
- Eychenne, R.; Chérel, M.; Haddad, F.; Guérard, F.; Gestin, J.F. Overview of the Most Promising Radionuclides for Targeted Alpha Therapy: The “Hopeful Eight”. Pharmaceutics 2021, 13, 906. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- Garin, E.; Tselikas, L.; Guiu, B.; Chalaye, J.; Edeline, J.; de Baere, T.; Assenat, E.; Tacher, V.; Robert, C.; Terroir-Cassou-Mounat, M.; et al. Personalised versus standard dosimetry approach of selective internal radiation therapy in patients with locally advanced hepatocellular carcinoma (DOSISPHERE-01): A randomised, multicentre, open-label phase 2 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, M.C.; de Geus, S.W.; Prevoo, H.A.; Hawinkels, L.J.; van de Velde, C.J.; Kuppen, P.J.; Vahrmeijer, A.L.; Sier, C.F. Selecting Targets for Tumor Imaging: An Overview of Cancer-Associated Membrane Proteins. Biomark. Cancer 2016, 8, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Aloj, L.; Attili, B.; Lau, D.; Caraco, C.; Lechermann, L.M.; Mendichovszky, I.A.; Harper, I.; Cheow, H.; Casey, R.T.; Sala, E.; et al. The emerging role of cell surface receptor and protein binding radiopharmaceuticals in cancer diagnostics and therapy. Nucl. Med. Biol. 2021, 92, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Das, A.; Bal, C.S. Tumor-targeting agents. In Nuclear Medicine and Immunology; Harsini, S., Alavi, A., Rezaei, N., Eds.; Springer Nature: Cham, Switzerland, 2022; pp. 217–236. [Google Scholar]
- Liolios, C.; Sachpekidis, C.; Schäfer, M.; Kopka, K. Bispecific radioligands targeting prostate-specific membrane antigen and gastrin-releasing peptide receptors on the surface of prostate cancer cells. J. Label. Comp. Radiopharm. 2019, 62, 510–522. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef]
- Khalaf, K.; Hana, D.; Chou, J.T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 656364. [Google Scholar] [CrossRef]
- Ni, Y.; Zhou, X.; Yang, J.; Shi, H.; Li, H.; Zhao, X.; Ma, X. The Role of Tumor-Stroma Interactions in Drug Resistance Within Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 637675. [Google Scholar] [CrossRef]
- Van der Heide, C.D.; Dalm, S.U. Radionuclide imaging and therapy directed towards the tumor microenvironment: A multi-cancer approach for personalized medicine. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4616–4641. [Google Scholar] [CrossRef] [PubMed]
- Kharaishvili, G.; Simkova, D.; Bouchalova, K.; Gachechiladze, M.; Narsia, N.; Bouchal, J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Corroyer-Dulmont, A.; Jaudet, C.; Frelin, A.M.; Fantin, J.; Weyts, K.; Vallis, K.A.; Falzone, N.; Sibson, N.R.; Chérel, M.; Kraeber-Bodéré, F.; et al. Radioimmunotherapy for Brain Metastases: The Potential for Inflammation as a Target of Choice. Front. Oncol. 2021, 11, 714514. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Sonanini, D.; Maurer, A.; Daldrup-Link, H.E. The yin and yang of imaging tumor associated macrophages with PET and MRI. Theranostics 2019, 9, 7730–7748. [Google Scholar] [CrossRef]
- Brummel, K.; Eerkens, A.L.; de Bruyn, M.; Nijman, H.W. Tumour-infiltrating lymphocytes: From prognosis to treatment selection. Br. J. Cancer 2023, 128, 451–458. [Google Scholar] [CrossRef]
- Masłowska, K.; Halik, P.K.; Tymecka, D.; Misicka, A.; Gniazdowska, E. The Role of VEGF Receptors as Molecular Target in Nuclear Medicine for Cancer Diagnosis and Combination Therapy. Cancers 2021, 13, 1072. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Lee, S.T.; Burvenich, I.; Scott, A.M. Novel Target Selection for Nuclear Medicine Studies. Semin. Nucl. Med. 2019, 49, 357–368. [Google Scholar] [CrossRef]
- Turck, R. Radio-pharmaceuticals for cancer treatment: Are they ready for prime time yet? Ann. Oncol. 2018, 29, 1594–1597. [Google Scholar] [CrossRef]
- Herrmann, K.; Schwaiger, M.; Lewis, J.S.; Solomon, S.B.; McNeil, B.J.; Baumann, M.; Gambhir, S.S.; Hricak, H.; Weissleder, R. Radiotheranostics: A roadmap for future development. Lancet Oncol. 2020, 21, e146–e156. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M. New developments in monoclonal antibodies for cancer detection and therapy. CA Cancer J. Clin. 1994, 44, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.C.; Buraggi, G.L. NATO Advanced Study Institute on “radiolabeled monoclonal antibodies for imaging and therapy—potential, problems, and prospects”. Int. J. Biol. Markers 1987, 2, 43–48. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- DeNardo, S.J.; DeNardo, G.L.; O’Grady, L.F.; Macey, D.J.; Mills, S.L.; Epstein, A.L.; Peng, J.S.; McGahan, J.P. Treatment of a patient with B cell lymphoma by I-131 LYM-1 monoclonal antibodies. Int. J. Biol. Markers 1987, 2, 49–53. [Google Scholar] [CrossRef]
- Morschhauser, F.; Radford, J.; Van Hoof, A.; Botto, B.; Rohatiner, A.Z.; Salles, G.; Soubeyran, P.; Tilly, H.; Bischof-Delaloye, A.; van Putten, W.L.; et al. 90Yttrium-ibritumomab tiuxetan consolidation of first remission in advanced-stage follicular non-Hodgkin lymphoma: Updated results after a median follow-up of 7.3 years from the International, Randomized, Phase III First-Line Indolent trial. J. Clin. Oncol. 2013, 31, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Kraeber-Bodéré, F.; Wegener, W.A.; Harousseau, J.L.; Petillon, M.O.; Huglo, D.; Trümper, L.H.; Meller, J.; Pfreundschuh, M.; Kirsch, C.M.; et al. High rates of durable responses with anti-CD22 fractionated radioimmunotherapy: Results of a multicenter, phase I/II study in non-Hodgkin’s lymphoma. J. Clin. Oncol. 2010, 28, 3709–3716. [Google Scholar] [CrossRef]
- Blakkisrud, J.; Løndalen, A.; Martinsen, A.C.; Dahle, J.; Holtedahl, J.E.; Bach-Gansmo, T.; Holte, H.; Kolstad, A.; Stokke, C. Tumor-Absorbed Dose for Non-Hodgkin Lymphoma Patients Treated with the Anti-CD37 Antibody Radionuclide Conjugate 177Lu-Lilotomab Satetraxetan. J. Nucl. Med. 2017, 58, 48–54. [Google Scholar] [CrossRef]
- Stokke, C.; Blakkisrud, J.; Løndalen, A.; Dahle, J.; Martinsen, A.C.T.; Holte, H.; Kolstad, A. Pre-dosing with lilotomab prior to therapy with 177Lu-lilotomab satetraxetan significantly increases the ratio of tumor to red marrow absorbed dose in non-Hodgkin lymphoma patients. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1233–1241. [Google Scholar] [CrossRef]
- Gritti, G.; Gianatti, A.; Petronzelli, F.; De Santis, R.; Pavoni, C.; Rossi, R.L.; Cattaneo, L.; Spagnoli, L.G.; Ferrari, S.; Rossi, A.; et al. Evaluation of tenascin-C by tenatumomab in T-cell non-Hodgkin lymphomas identifies a new target for radioimmunotherapy. Oncotarget 2018, 9, 9766–9775. [Google Scholar] [CrossRef]
- Buchsbaum, D.J. Experimental tumor targeting with radiolabeled ligands. Cancer 1997, 80, 2371–2377. [Google Scholar] [CrossRef]
- Bäck, T.; Haraldsson, B.; Hultborn, R.; Jensen, H.; Johansson, M.E.; Lindegren, S.; Jacobsson, L. Glomerular filtration rate after alpha-radioimmunotherapy with 211At-MX35-F(ab′)2: A long-term study of renal function in nude mice. Cancer Biother. Radiopharm. 2009, 24, 649–658. [Google Scholar] [PubMed]
- Adams, G.P.; Shaller, C.C.; Chappell, L.L.; Wu, C.; Horak, E.M.; Simmons, H.H.; Litwin, S.; Marks, J.D.; Weiner, L.M.; Brechbiel, M.W. Delivery of the alpha-emitting radioisotope bismuth-213 to solid tumors via single-chain Fv and diabody molecules. Nucl. Med. Biol. 2000, 27, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Diebolder, P.; Mpoy, C.; Scott, J.; Huynh, T.T.; Fields, R.; Spitzer, D.; Bandara, N.; Rogers, B.E. Preclinical Evaluation of an Engineered Single-Chain Fragment Variable-Fragment Crystallizable Targeting Human CD44. J. Nucl. Med. 2021, 62, 137–143. [Google Scholar] [CrossRef]
- Altunay, B.; Morgenroth, A.; Beheshti, M.; Vogg, A.; Wong, N.C.L.; Ting, H.H.; Biersack, H.J.; Stickeler, E.; Mottaghy, F.M. HER2-directed antibodies, affibodies and nanobodies as drug-delivery vehicles in breast cancer with a specific focus on radioimmunotherapy and radioimmunoimaging. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1371–1389. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, V.; Orlova, A.; Pehrson, R.; Galli, J.; Baastrup, B.; Andersson, K.; Sandström, M.; Rosik, D.; Carlsson, J.; Lundqvist, H.; et al. Radionuclide therapy of HER2-positive microxenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res. 2007, 67, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Barbet, J.; Kraeber-Bodéré, F.; Vuillez, J.P.; Gautherot, E.; Rouvier, E.; Chatal, J.F. Pretargeting with the affinity enhancement system for radioimmunotherapy. Cancer Biother. Radiopharm. 1999, 14, 153–166. [Google Scholar] [CrossRef]
- Kraeber-Bodéré, F.; Rousseau, C.; Bodet-Milin, C.; Ferrer, L.; Faivre-Chauvet, A.; Campion, L.; Vuillez, J.P.; Devillers, A.; Chang, C.H.; Goldenberg, D.M.; et al. Targeting, toxicity, and efficacy of 2-step, pretargeted radioimmunotherapy using a chimeric bispecific antibody and 131I-labeled bivalent hapten in a phase I optimization clinical trial. J. Nucl. Med. 2006, 47, 247–255. [Google Scholar]
- Salaun, P.Y.; Campion, L.; Bournaud, C.; Faivre-Chauvet, A.; Vuillez, J.P.; Taieb, D.; Ansquer, C.; Rousseau, C.; Borson-Chazot, F.; Bardet, S.; et al. Phase II trial of anticarcinoembryonic antigen pretargeted radioimmunotherapy in progressive metastatic medullary thyroid carcinoma: Biomarker response and survival improvement. J. Nucl. Med. 2012, 53, 1185–1192. [Google Scholar] [CrossRef]
- DeNardo, G.L.; Schlom, J.; Buchsbaum, D.J.; Meredith, R.F.; O’Donoghue, J.A.; Sgouros, G.; Humm, J.L.; DeNardo, S.J. Rationales, evidence, and design considerations for fractionated radioimmunotherapy. Cancer 2002, 94, 1332–1348. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Vallabhajosula, S.; Christos, P.J.; Jhanwar, Y.S.; Batra, J.S.; Lam, L.; Osborne, J.; Beltran, H.; Molina, A.M.; Goldsmith, S.J.; et al. Phase 1/2 study of fractionated dose lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 (177 Lu-J591) for metastatic castration-resistant prostate cancer. Cancer 2019, 125, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Kraeber-Bodere, F.; Pallardy, A.; Maisonneuve, H.; Campion, L.; Moreau, A.; Soubeyran, I.; Le Gouill, S.; Tournilhac, O.; Daguindau, E.; Jardel, H.; et al. Consolidation anti-CD22 fractionated radioimmunotherapy with 90Y-epratuzumab tetraxetan following R-CHOP in elderly patients with diffuse large B-cell lymphoma: A prospective, single group, phase 2 trial. Lancet Haematol. 2017, 4, e35–e45. [Google Scholar] [CrossRef]
- White, J.M.; Escorcia, F.E.; Viola, N.T. Perspectives on metals-based radioimmunotherapy (RIT): Moving forward. Theranostics 2021, 11, 6293–6314. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, U.; Cremonesi, M.; Lassmann, M. Individualized Dosimetry for Theranostics: Necessary, Nice to Have, or Counterproductive? J. Nucl. Med. 2017, 58, 97S–103S. [Google Scholar] [CrossRef] [PubMed]
- Filippi, L.; Bagni, O.; Nervi, C. Aptamer-based technology for radionuclide targeted imaging and therapy: A promising weapon against cancer. Expert Rev. Med. Devices 2020, 17, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Gijs, M.; Aerts, A.; Impens, N.; Baatout, S.; Luxen, A. Aptamers as radiopharmaceuticals for nuclear imaging and therapy. Nucl. Med. Biol. 2016, 43, 253–271. [Google Scholar] [CrossRef]
- Varmira, K.; Hosseinimehr, S.J.; Noaparast, Z.; Abedi, S.M. An improved radiolabelled RNA aptamer molecule for HER2 imaging in cancers. J. Drug Target 2014, 22, 116–122. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef]
- Zhong, Y.; Wu, P.; He, J.; Zhong, L.; Zhao, Y. Advances of aptamer-based clinical applications for the diagnosis and therapy of cancer. Discov. Med. 2020, 29, 169–180. [Google Scholar]
- De Visser, M.; Verwijnen, S.M.; de Jong, M. Update: Improvement strategies for peptide receptor scintigraphy and radionuclide therapy. Cancer Biother. Radiopharm. 2008, 23, 137–157. [Google Scholar]
- Tornesello, A.L.; Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules 2017, 22, 1282. [Google Scholar] [CrossRef]
- Abbasi Gharibkandi, N.; Conlon, J.M.; Hosseinimehr, S.J. Strategies for improving stability and pharmacokinetic characteristics of radiolabeled peptides for imaging and therapy. Peptides 2020, 133, 170385. [Google Scholar] [CrossRef] [PubMed]
- Qu, T.; Wang, Y.; Zhu, Z.; Rusckowski, M.; Hnatowich, D.J. Different chelators and different peptides together influence the in vitro and mouse in vivo properties of 99Tcm. Nucl. Med. Commun. 2001, 22, 203–215. [Google Scholar] [CrossRef]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: The chelate makes the difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Weiner, R.E.; Thakur, M.L. Radiolabeled peptides in the diagnosis and therapy of oncological diseases. Appl. Radiat. Isot. 2002, 57, 749–763. [Google Scholar] [CrossRef]
- Morgat, C.; Mishra, A.K.; Varshney, R.; Allard, M.; Fernandez, P.; Hindié, E. Targeting neuropeptide receptors for cancer imaging and therapy: Perspectives with bombesin, neurotensin, and neuropeptide-Y receptors. J. Nucl. Med. 2014, 55, 1650–1657. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. An Overview of Bioactive Peptides for in vivo Imaging and Therapy in Human Diseases. Mini Rev. Med. Chem. 2017, 17, 758–770. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Schaer, J.C.; Laissue, J.A. Somatostatin receptor sst1-sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur. J. Nucl. Med. 2001, 28, 836–846. [Google Scholar] [CrossRef]
- Eychenne, R.; Bouvry, C.; Bourgeois, M.; Loyer, P.; Benoist, E.; Lepareur, N. Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy. Molecules 2020, 25, 4012. [Google Scholar] [CrossRef]
- Hennrich, U.; Benešová, M. [68Ga]Ga-DOTA-TOC: The First FDA-Approved 68Ga-Radiopharmaceutical for PET Imaging. Pharmaceuticals 2020, 13, 38. [Google Scholar] [CrossRef]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Kaltsas, G.A.; Papadogias, D.; Makras, P.; Grossman, A.B. Treatment of advanced neuroendocrine tumours with radiolabelled somatostatin analogues. Endocr. Relat. Cancer 2005, 12, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Das, S.; Al-Toubah, T.; Pelle, E.; El-Haddad, G.; Strosberg, J. Somatostatin receptor radionuclide therapy in neuroendocrine tumors. Endocr. Relat. Cancer 2021, 28, R81–R93. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, L.; Boschi, A.; Cittanti, C.; Martini, P.; Panareo, S.; Tonini, E.; Nieri, A.; Urso, L.; Caracciolo, M.; Lodi, L.; et al. 90Y/177Lu-DOTATOC: From Preclinical Studies to Application in Humans. Pharmaceutics 2021, 13, 1463. [Google Scholar] [CrossRef] [PubMed]
- Puliani, G.; Chiefari, A.; Mormando, M.; Bianchini, M.; Lauretta, R.; Appetecchia, M. New Insights in PRRT: Lessons from 2021. Front. Endocrinol. 2022, 13, 861434. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. NETTER-1 Trial Investigators. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Ahmadi Bidakhvidi, N.; Goffin, K.; Dekervel, J.; Baete, K.; Nackaerts, K.; Clement, P.; Van Cutsem, E.; Verslype, C.; Deroose, C.M. Peptide Receptor Radionuclide Therapy Targeting the Somatostatin Receptor: Basic Principles, Clinical Applications and Optimization Strategies. Cancers 2021, 14, 129. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. ²¹³Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106–2119. [Google Scholar] [CrossRef]
- Ballal, S.; Yadav, M.P.; Bal, C.; Sahoo, R.K.; Tripathi, M. Broadening horizons with 225Ac-DOTATATE targeted alpha therapy for gastroenteropancreatic neuroendocrine tumour patients stable or refractory to 177Lu-DOTATATE PRRT: First clinical experience on the efficacy and safety. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 934–946. [Google Scholar] [CrossRef]
- Koh, T.T.; Bezak, E.; Chan, D.; Cehic, G. Targeted alpha-particle therapy in neuroendocrine neoplasms: A systematic review. World J. Nucl. Med. 2021, 20, 329–335. [Google Scholar] [CrossRef]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Mäcke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Peitl, P.K.; Velikyan, I. Current Status of Radiopharmaceuticals for the Theranostics of Neuroendocrine Neoplasms. Pharmaceuticals 2017, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Fani, M.; Behe, M.; Brink, I.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; Weber, W.A. First clinical evidence that imaging with somatostatin receptor antagonists is feasible. J. Nucl. Med. 2011, 52, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Nicolas, G.P.; Wild, D. Somatostatin Receptor Antagonists for Imaging and Therapy. J. Nucl. Med. 2017, 58, 61S–66S. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Fani, M.; Fischer, R.; Del Pozzo, L.; Kaul, F.; Krebs, S.; Fischer, R.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; et al. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: A pilot study. J. Nucl. Med. 2014, 55, 1248–1252. [Google Scholar] [CrossRef]
- Reidy-Lagunes, D.; Pandit-Taskar, N.; O’Donoghue, J.A.; Krebs, S.; Staton, K.D.; Lyashchenko, S.K.; Lewis, J.S.; Raj, N.; Gönen, M.; Lohrmann, C.; et al. Phase I Trial of Well-Differentiated Neuroendocrine Tumors (NETs) with Radiolabeled Somatostatin Antagonist 177Lu-Satoreotide Tetraxetan. Clin. Cancer Res. 2019, 25, 6939–6947. [Google Scholar] [CrossRef]
- Baum, R.P.; Zhang, J.; Schuchardt, C.; Müller, D.; Mäcke, H. First-in-Humans Study of the SSTR Antagonist 177Lu-DOTA-LM3 for Peptide Receptor Radionuclide Therapy in Patients with Metastatic Neuroendocrine Neoplasms: Dosimetry, Safety, and Efficacy. J. Nucl. Med. 2021, 62, 1571–1581. [Google Scholar] [CrossRef]
- Fani, M.; Mansi, R.; Nicolas, G.P.; Wild, D. Radiolabeled Somatostatin Analogs-A Continuously Evolving Class of Radiopharmaceuticals. Cancers 2022, 14, 1172. [Google Scholar] [CrossRef]
- Harris, P.E.; Zhernosekov, K. The evolution of PRRT for the treatment of neuroendocrine tumors; What comes next? Front. Endocrinol. 2022, 13, 941832. [Google Scholar] [CrossRef]
- Van de Wiele, C.; Dumont, F.; van Belle, S.; Slegers, G.; Peers, S.H.; Dierckx, R.A. Is there a role for agonist gastrin-releasing peptide receptor radioligands in tumour imaging? Nucl. Med. Commun. 2001, 22, 5–15. [Google Scholar] [CrossRef]
- Smith, C.J.; Volkert, W.A.; Hoffman, T.J. Radiolabeled peptide conjugates for targeting of the bombesin receptor superfamily subtypes. Nucl. Med. Biol. 2005, 32, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Nock, B.A.; Dalm, S.U.; Busstra, M.B.; van Weerden, W.M.; Maina, T. Radiolabeled Bombesin Analogs. Cancers 2021, 13, 5766. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Ferrari, M.; Nunn, A.; Llull, J.; Cremonesi, M.; Martano, L.; Laurora, G.; Scardino, E.; Tiberini, S.; Bufi, G.; et al. 177Lu-AMBA bombesin analogue in hormone refractory prostate cancer patients: A phase I escalation study with single-cycle administrations. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, S221. [Google Scholar]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef]
- Maina, T.; Nock, B.A.; Kulkarni, H.; Singh, A.; Baum, R.P. Theranostic Prospects of Gastrin-Releasing Peptide Receptor-Radioantagonists in Oncology. PET Clin. 2017, 12, 297–309. [Google Scholar] [CrossRef]
- Kurth, J.; Krause, B.J.; Schwarzenböck, S.M.; Bergner, C.; Hakenberg, O.W.; Heuschkel, M. First-in-human dosimetry of gastrin-releasing peptide receptor antagonist [177Lu]Lu-RM2: A radiopharmaceutical for the treatment of metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 123–135. [Google Scholar] [CrossRef]
- Günther, T.; Deiser, S.; Felber, V.; Beck, R.; Wester, H.J. Substitution of l-Tryptophan by α-Methyl-l-Tryptophan in 177Lu-RM2 Results in 177Lu-AMTG, a High-Affinity Gastrin-Releasing Peptide Receptor Ligand with Improved In Vivo Stability. J. Nucl. Med. 2022, 63, 1364–1370. [Google Scholar] [CrossRef]
- Montemagno, C.; Raes, F.; Ahmadi, M.; Bacot, S.; Debiossat, M.; Leenhardt, J.; Boutonnat, J.; Orlandi, F.; Barbato, D.; Tedesco, M.; et al. In Vivo Biodistribution and Efficacy Evaluation of NeoB, a Radiotracer Targeted to GRPR, in Mice Bearing Gastrointestinal Stromal Tumor. Cancers 2021, 13, 1051. [Google Scholar] [CrossRef]
- Ruigrok, E.A.M.; Verhoeven, M.; Konijnenberg, M.W.; de Blois, E.; de Ridder, C.M.A.; Stuurman, D.C.; Bertarione, L.; Rolfo, K.; de Jong, M.; Dalm, S.U. Safety of [177Lu]Lu-NeoB treatment: A preclinical study characterizing absorbed dose and acute, early, and late organ toxicity. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4440–4451. [Google Scholar] [CrossRef]
- Huynh, T.T.; van Dam, E.M.; Sreekumar, S.; Mpoy, C.; Blyth, B.J.; Muntz, F.; Harris, M.J.; Rogers, B.E. Copper-67-Labeled Bombesin Peptide for Targeted Radionuclide Therapy of Prostate Cancer. Pharmaceuticals 2022, 15, 728. [Google Scholar] [CrossRef]
- Majkowska-Pilip, A.; Halik, P.K.; Gniazdowska, E. The Significance of NK1 Receptor Ligands and Their Application in Targeted Radionuclide Tumour Therapy. Pharmaceutics 2019, 11, 443. [Google Scholar] [CrossRef] [PubMed]
- Kneifel, S.; Cordier, D.; Good, S.; Ionescu, M.C.; Ghaffari, A.; Hofer, S.; Kretzschmar, M.; Tolnay, M.; Apostolidis, C.; Waser, B.; et al. Local targeting of malignant gliomas by the diffusible peptidic vector 1,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid-substance p. Clin. Cancer Res. 2006, 12, 3843–3850. [Google Scholar] [CrossRef] [PubMed]
- Cordier, D.; Krolicki, L.; Morgenstern, A.; Merlo, A. Targeted Radiolabeled Compounds in Glioma Therapy. Semin. Nucl. Med. 2016, 46, 243–249. [Google Scholar] [CrossRef]
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Prolonged survival in secondary glioblastoma following local injection of targeted alpha therapy with 213Bi-substance P analogue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1636–1644. [Google Scholar] [CrossRef]
- Królicki, L.; Kunikowska, J.; Bruchertseifer, F.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Rola, R.; Morgenstern, A.; Rosiak, E.; et al. 225Ac- and 213Bi-Substance P Analogues for Glioma Therapy. Semin. Nucl. Med. 2020, 50, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Halik, P.K.; Lipiński, P.F.J.; Matalińska, J.; Koźmiński, P.; Misicka, A.; Gniazdowska, E. Radiochemical Synthesis and Evaluation of Novel Radioconjugates of Neurokinin 1 Receptor Antagonist Aprepitant Dedicated for NK1R-Positive Tumors. Molecules 2020, 25, 3756. [Google Scholar] [CrossRef] [PubMed]
- Matalińska, J.; Kosińska, K.; Halik, P.K.; Koźmiński, P.; Lipiński, P.F.J.; Gniazdowska, E.; Misicka, A. Novel NK1R-Targeted 68Ga-/177Lu-Radioconjugates with Potential Application against Glioblastoma Multiforme: Preliminary Exploration of Structure-Activity Relationships. Int. J. Mol. Sci. 2022, 23, 1214. [Google Scholar] [CrossRef]
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413. [Google Scholar] [CrossRef]
- Ansquer, C.; Kraeber-Bodéré, F.; Chatal, J.F. Current status and perspectives in peptide receptor radiation therapy. Curr. Pharm. Des. 2009, 15, 2453–2462. [Google Scholar] [CrossRef]
- Franco Machado, J.; Silva, R.D.; Melo, R.; Correia, J.D.G. Less Exploited GPCRs in Precision Medicine: Targets for Molecular Imaging and Theranostics. Molecules 2018, 24, 49. [Google Scholar] [CrossRef]
- Thakur, M.L.; Tripathi, S.K.; Gomella, L.G.; Salmanoglu, E.; Kim, S.; Kelly, W.K.; Keith, S.W.; Intenzo, C.; McCue, P.; Hoffman-Censits, J.; et al. Imaging urothelial bladder cancer: A VPAC PET targeted approach. Can. J. Urol. 2021, 28, 10596–10602. [Google Scholar] [PubMed]
- Von Guggenberg, E.; Kolenc, P.; Rottenburger, C.; Mikołajczak, R.; Hubalewska-Dydejczyk, A. Update on Preclinical Development and Clinical Translation of Cholecystokinin-2 Receptor Targeting Radiopharmaceuticals. Cancers 2021, 13, 5776. [Google Scholar] [CrossRef] [PubMed]
- Rottenburger, C.; Nicolas, G.P.; McDougall, L.; Kaul, F.; Cachovan, M.; Vija, A.H.; Schibli, R.; Geistlich, S.; Schumann, A.; Rau, T.; et al. Cholecystokinin 2 Receptor Agonist 177Lu-PP-F11N for Radionuclide Therapy of Medullary Thyroid Carcinoma: Results of the Lumed Phase 0a Study. J. Nucl. Med. 2020, 61, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, N.; Zhang, T.T.; Nakanishi, T. Involvement of CXCR4 in Normal and Abnormal Development. Cells 2019, 8, 185. [Google Scholar] [CrossRef]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar]
- Werner, R.A.; Kircher, S.; Higuchi, T.; Kircher, M.; Schirbel, A.; Wester, H.J.; Buck, A.K.; Pomper, M.G.; Rowe, S.P.; Lapa, C. CXCR4-Directed Imaging in Solid Tumors. Front. Oncol. 2019, 9, 770. [Google Scholar] [CrossRef]
- Kircher, M.; Herhaus, P.; Schottelius, M.; Buck, A.K.; Werner, R.A.; Wester, H.J.; Keller, U.; Lapa, C. CXCR4-directed theranostics in oncology and inflammation. Ann. Nucl. Med. 2018, 32, 503–511. [Google Scholar] [CrossRef]
- Schottelius, M.; Osl, T.; Poschenrieder, A.; Hoffmann, F.; Beykan, S.; Hänscheid, H.; Schirbel, A.; Buck, A.K.; Kropf, S.; Schwaiger, M.; et al. [177Lu]pentixather: Comprehensive Preclinical Characterization of a First CXCR4-directed Endoradiotherapeutic Agent. Theranostics 2017, 7, 2350–2362. [Google Scholar] [CrossRef]
- Osl, T.; Schmidt, A.; Schwaiger, M.; Schottelius, M.; Wester, H.J. A new class of PentixaFor- and PentixaTher-based theranostic agents with enhanced CXCR4-targeting efficiency. Theranostics 2020, 10, 8264–8280. [Google Scholar] [CrossRef]
- Herrmann, K.; Schottelius, M.; Lapa, C.; Osl, T.; Poschenrieder, A.; Hänscheid, H.; Lückerath, K.; Schreder, M.; Bluemel, C.; Knott, M.; et al. First-in-Human Experience of CXCR4-Directed Endoradiotherapy with 177Lu- and 90Y-Labeled Pentixather in Advanced-Stage Multiple Myeloma with Extensive Intra- and Extramedullary Disease. J. Nucl. Med. 2016, 57, 248–251. [Google Scholar] [CrossRef]
- Maurer, S.; Herhaus, P.; Lippenmeyer, R.; Hänscheid, H.; Kircher, M.; Schirbel, A.; Maurer, H.C.; Buck, A.K.; Wester, H.J.; Einsele, H.; et al. Side Effects of CXC-Chemokine Receptor 4-Directed Endoradiotherapy with Pentixather Before Hematopoietic Stem Cell Transplantation. J. Nucl. Med. 2019, 60, 1399–1405. [Google Scholar] [CrossRef]
- Schottelius, M.; Herrmann, K.; Lapa, C. In Vivo Targeting of CXCR4-New Horizons. Cancers 2021, 13, 5920. [Google Scholar] [CrossRef]
- Buck, A.K.; Serfling, S.E.; Lindner, T.; Hänscheid, H.; Schirbel, A.; Hahner, S.; Fassnacht, M.; Einsele, H.; Werner, R.A. CXCR4-targeted theranostics in oncology. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4133–4144. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xue, Q.; Yao, S. Nuclear Medicine Application of Pentixafor/Pentixather Targeting CXCR4 for Imaging and Therapy in Related Disease. Mini Rev. Med. Chem. 2023, 23, 787–803. [Google Scholar] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Decristoforo, C. Radiolabelled RGD peptides and peptidomimetics for tumour targeting. Front. Biosci. 2009, 14, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Notni, J. RGD Forever!—Past, Present, and Future of a 3-Letter-Code in Radiopharmacy and Life Sciences. Pharmaceuticals 2023, 16, 56. [Google Scholar] [CrossRef]
- Kossatz, S.; Beer, A.J.; Notni, J. It’s Time to Shift the Paradigm: Translation and Clinical Application of Non-αvβ3 Integrin Targeting Radiopharmaceuticals. Cancers 2021, 13, 5958. [Google Scholar] [CrossRef]
- Masłowska, K.; Witkowska, E.; Tymecka, D.; Halik, P.K.; Misicka, A.; Gniazdowska, E. Synthesis, Physicochemical and Biological Study of Gallium-68- and Lutetium-177-Labeled VEGF-A165/NRP-1 Complex Inhibitors Based on Peptide A7R and Branched Peptidomimetic. Pharmaceutics 2022, 14, 100. [Google Scholar] [CrossRef]
- Ayo, A.; Laakkonen, P. Peptide-Based Strategies for Targeted Tumor Treatment and Imaging. Pharmaceutics 2021, 13, 481. [Google Scholar] [CrossRef]
- Khalily, M.P.; Soydan, M. Peptide-based diagnostic and therapeutic agents: Where we are and where we are heading? Chem. Biol. Drug Des. 2023, 101, 772–793. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Liu, H.; Cheng, Z. Radiolabeled Peptide Probes for Liver Cancer Imaging. Curr. Med. Chem. 2020, 27, 6968–6986. [Google Scholar] [CrossRef] [PubMed]
- Ciobanasu, C. Peptides-based therapy and diagnosis. Strategies for non-invasive therapies in cancer. J. Drug Target 2021, 29, 1063–1079. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.P.; Singh, A.; Schuchardt, C.; Kulkarni, H.R.; Klette, I.; Wiessalla, S.; Osterkamp, F.; Reineke, U.; Smerling, C. 177Lu-3BP-227 for Neurotensin Receptor 1-Targeted Therapy of Metastatic Pancreatic Adenocarcinoma: First Clinical Results. J. Nucl. Med. 2018, 59, 809–814. [Google Scholar] [CrossRef]
- Nock, B.A.; Kanellopoulos, P.; Chepurny, O.G.; Rouchota, M.; Loudos, G.; Holz, G.G.; Krenning, E.P.; Maina, T. Nonpeptidic Z360-Analogs Tagged with Trivalent Radiometals as Anti-CCK2R Cancer Theranostic Agents: A Preclinical Study. Pharmaceutics 2022, 14, 666. [Google Scholar] [CrossRef]
- Gdowski, A.S.; Ranjan, A.; Vishwanatha, J.K. Current concepts in bone metastasis, contemporary therapeutic strategies and ongoing clinical trials. J. Exp. Clin. Cancer Res. 2017, 36, 108. [Google Scholar] [CrossRef]
- Liepe, K.; Kotzerke, J. Internal radiotherapy of painful bone metastases. Methods 2011, 55, 258–270. [Google Scholar] [CrossRef]
- Potsaid, M.S.; Irwin, R.J., Jr.; Castronovo, F.P.; Prout, G.R., Jr.; Harvey, W.J.; Francis, M.D.; Tofe, A.J.; Zamenhof, R.G. [32P] diphosphonate dose determination in patients with bone metastases from prostatic carcinoma. J. Nucl. Med. 1978, 19, 98–104. [Google Scholar]
- Roberts, D.J., Jr. 32P-sodium phosphate treatment of metastatic malignant disease. Clin. Nucl. Med. 1979, 4, 92–93. [Google Scholar] [CrossRef]
- Mathieu, L.; Chevalier, P.; Galy, G.; Berger, M. Preparation of rhenium-186 labelled EHDP and its possible use in the treatment of osseous neoplasms. Int. J. Appl. Radiat. Isot. 1979, 30, 725–727. [Google Scholar] [CrossRef]
- Hosain, F.; Spencer, R.P. Radiopharmaceuticals for palliation of metastatic osseous lesions: Biologic and physical background. Semin. Nucl. Med. 1992, 22, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Bouchet, L.G.; Bolch, W.E.; Goddu, S.M.; Howell, R.W.; Rao, D.V. Considerations in the selection of radiopharmaceuticals for palliation of bone pain from metastatic osseous lesions. J. Nucl. Med. 2000, 41, 682–687. [Google Scholar] [PubMed]
- Pauwels, E.K.; Stokkel, M.P. Radiopharmaceuticals for bone lesions. Imaging and therapy in clinical practice. Q. J. Nucl. Med. 2001, 45, 18–26. [Google Scholar] [PubMed]
- Lange, R.; Ter Heine, R.; Knapp, F.F.; de Klerk, J.M.; Bloemendal, H.J.; Hendrikse, N.H. Pharmaceutical and clinical development of phosphonate-based radiopharmaceuticals for the targeted treatment of bone metastases. Bone 2016, 91, 159–179. [Google Scholar] [CrossRef]
- Liepe, K.; Shinto, A. From palliative therapy to prolongation of survival: 223RaCl2 in the treatment of bone metastases. Ther. Adv. Med. Oncol. 2016, 8, 294–304. [Google Scholar] [CrossRef]
- Das, T.; Banerjee, S. Radiopharmaceuticals for metastatic bone pain palliation: Available options in the clinical domain and their comparisons. Clin. Exp. Metastasis 2017, 34, 1–10. [Google Scholar] [CrossRef]
- Fernández, R.; Eppard, E.; Lehnert, W.; Jiménez-Franco, L.D.; Soza-Ried, C.; Ceballos, M.; Ribbeck, J.; Kluge, A.; Rösch, F.; Meckel, M.; et al. Evaluation of Safety and Dosimetry of 177Lu-DOTA-ZOL for Therapy of Bone Metastases. J. Nucl. Med. 2021, 62, 1126–1132. [Google Scholar] [CrossRef]
- Palmedo, H.; Manka-Waluch, A.; Albers, P.; Schmidt-Wolf, I.G.; Reinhardt, M.; Ezziddin, S.; Joe, A.; Roedel, R.; Fimmers, R.; Knapp, F.F., Jr.; et al. Repeated bone-targeted therapy for hormone-refractory prostate carcinoma: Tandomized phase II trial with the new, high-energy radiopharmaceutical rhenium-188 hydroxyethylidenediphosphonate. J. Clin. Oncol. 2003, 21, 2869–2875. [Google Scholar] [CrossRef]
- Biersack, H.J.; Palmedo, H.; Andris, A.; Rogenhofer, S.; Knapp, F.F.; Guhlke, S.; Ezziddin, S.; Bucerius, J.; von Mallek, D. Palliation and survival after repeated 188Re-HEDP therapy of hormone-refractory bone metastases of prostate cancer: A retrospective analysis. J. Nucl. Med. 2011, 52, 172–1726. [Google Scholar] [CrossRef]
- Guerra-Liberal, F.D.C.; Tavares, A.A.S.; Tavares, J.M.R.S. Palliative treatment of metastatic bone pain with radiopharmaceuticals: A perspective beyond Strontium-89 and Samarium-153. Appl. Radiat. Isot. 2016, 110, 87–99. [Google Scholar] [CrossRef]
- Liepe, K.; Murray, I.; Flux, G. Dosimetry of Bone Seeking Beta Emitters for Bone Pain Palliation Metastases. Semin. Nucl. Med. 2022, 52, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Askari, E.; Harsini, S.; Vahidfar, N.; Divband, G.; Sadeghi, R. 177Lu-EDTMP for Metastatic Bone Pain Palliation: A Systematic Review and Meta-Analysis. Cancer Biother. Radiopharm. 2021, 36, 383–390. [Google Scholar] [CrossRef]
- Wieland, D.M.; Swanson, D.P.; Brown, L.E.; Beierwaltes, W.H. Imaging the adrenal medulla with an I-131-labeled antiadrenergic agent. J. Nucl. Med. 1979, 20, 155–158. [Google Scholar]
- Vallabhajosula, S.; Nikolopoulou, A. Radioiodinated metaiodobenzylguanidine (MIBG): Radiochemistry, biology, and pharmacology. Semin. Nucl. Med. 2011, 41, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Erwin, W.; Chasen, B. Targeted Radionuclide Therapy for Patients with Metastatic Pheochromocytoma and Paraganglioma: From Low-Specific-Activity to High-Specific-Activity Iodine-131 Metaiodobenzylguanidine. Cancers 2019, 11, 1018. [Google Scholar] [CrossRef] [PubMed]
- Jungels, C.; Karfis, I. 131I-metaiodobenzylguanidine and peptide receptor radionuclide therapy in pheochromocytoma and paraganglioma. Curr. Opin. Oncol. 2021, 33, 33–39. [Google Scholar] [CrossRef]
- Wilson, J.S.; Gains, J.E.; Moroz, V.; Wheatley, K.; Gaze, M.N. A systematic review of 131I-meta iodobenzylguanidine molecular radiotherapy for neuroblastoma. Eur. J. Cancer 2014, 50, 801–815. [Google Scholar] [CrossRef]
- Giammarile, F.; Chiti, A.; Lassmann, M.; Brans, B.; Flux, G. EANM procedure guidelines for 131I-meta-iodobenzylguanidine (131I-mIBG) therapy. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1039–1047. [Google Scholar] [CrossRef]
- French, S.; DuBois, S.G.; Horn, B.; Granger, M.; Hawkins, R.; Pass, A.; Plummer, E.; Matthay, K. 131I-MIBG followed by consolidation with busulfan, melphalan and autologous stem cell transplantation for refractory neuroblastoma. Pediatr. Blood Cancer 2013, 60, 879–884. [Google Scholar] [CrossRef]
- DuBois, S.G.; Chesler, L.; Groshen, S.; Hawkins, R.; Goodarzian, F.; Shimada, H.; Yanik, G.; Tagen, M.; Stewart, C.; Mosse, Y.P.; et al. Phase I study of vincristine, irinotecan, and ¹³¹I-metaiodobenzylguanidine for patients with relapsed or refractory neuroblastoma: A new approaches to neuroblastoma therapy trial. Clin. Cancer Res. 2012, 18, 2679–2686. [Google Scholar] [CrossRef]
- Zhang, X.; Wakabayashi, H.; Hiromasa, T.; Kayano, D.; Kinuya, S. Recent Advances in Radiopharmaceutical Theranostics of Pheochromocytoma and Paraganglioma. Semin. Nucl. Med. 2023, 53, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Prado-Wohlwend, S.; Del Olmo-García, M.I.; Bello-Arques, P.; Merino-Torres, J.F. Response to targeted radionuclide therapy with [131I]MIBG AND [177Lu]Lu-DOTA-TATE according to adrenal vs. extra-adrenal primary location in metastatic paragangliomas and pheochromocytomas: A systematic review. Front. Endocrinol. 2022, 13, 957172. [Google Scholar] [CrossRef] [PubMed]
- Pryma, D.A.; Chin, B.B.; Noto, R.B.; Dillon, J.S.; Perkins, S.; Solnes, L.; Kostakoglu, L.; Serafini, A.N.; Pampaloni, M.H.; Jensen, J.; et al. Efficacy and Safety of High-Specific-Activity 131I-MIBG Therapy in Patients with Advanced Pheochromocytoma or Paraganglioma. J. Nucl. Med. 2019, 60, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Sisson, J.C.; Shapiro, B.; Hutchinson, R.J.; Shulkin, B.L.; Zempel, S. Survival of patients with neuroblastoma treated with 125-I MIBG. Am. J. Clin. Oncol. 1996, 19, 144–148. [Google Scholar] [CrossRef]
- Ohshima, Y.; Sudo, H.; Watanabe, S.; Nagatsu, K.; Tsuji, A.B.; Sakashita, T.; Ito, Y.M.; Yoshinaga, K.; Higashi, T.; Ishioka, N.S. Antitumor effects of radionuclide treatment using α-emitting meta-211At-astato-benzylguanidine in a PC12 pheochromocytoma model. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Ukon, N.; Higashi, T.; Hosono, M.; Kinuya, S.; Yamada, T.; Yanagida, S.; Namba, M.; Nakamura, Y. Manual on the proper use of meta-[211At] astato-benzylguanidine ([211At] MABG) injections in clinical trials for targeted alpha therapy (1st edition). Ann. Nucl. Med. 2022, 36, 695–709. [Google Scholar] [CrossRef]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Backhaus, P.; Noto, B.; Avramovic, N.; Grubert, L.S.; Huss, S.; Bögemann, M.; Stegger, L.; Weckesser, M.; Schäfers, M.; Rahbar, K. Targeting PSMA by radioligands in non-prostate disease-current status and future perspectives. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 860–877. [Google Scholar] [CrossRef]
- Uijen, M.J.M.; Derks, Y.H.W.; Merkx, R.I.J.; Schilham, M.G.M.; Roosen, J.; Privé, B.M.; van Lith, S.A.M.; van Herpen, C.M.L.; Gotthardt, M.; Heskamp, S.; et al. PSMA radioligand therapy for solid tumors other than prostate cancer: Background, opportunities, challenges, and first clinical reports. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 4350–4368. [Google Scholar] [CrossRef]
- An, S.; Huang, G.; Liu, J.; Wei, W. PSMA-targeted theranostics of solid tumors: Applications beyond prostate cancers. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3973–3976. [Google Scholar] [CrossRef]
- Kratochwil, C.; Haberkorn, U.; Giesel, F.L. Radionuclide Therapy of Metastatic Prostate Cancer. Semin. Nucl. Med. 2019, 49, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, H.R.; Singh, A.; Schuchardt, C.; Niepsch, K.; Sayeg, M.; Leshch, Y.; Wester, H.J.; Baum, R.P. PSMA-Based Radioligand Therapy for Metastatic Castration-Resistant Prostate Cancer: The Bad Berka Experience Since 2013. J. Nucl. Med. 2016, 57, 97S–104S. [Google Scholar] [CrossRef]
- Sanli, Y.; Simsek, D.H.; Sanli, O.; Subramaniam, R.M.; Kendi, A.T. 177Lu-PSMA Therapy in Metastatic Castration-Resistant Prostate Cancer. Biomedicines 2021, 9, 430. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, K.; Ahmadzadehfar, H.; Kratochwil, C.; Haberkorn, U.; Schäfers, M.; Essler, M.; Baum, R.P.; Kulkarni, H.R.; Schmidt, M.; Drzezga, A.; et al. German Multicenter Study Investigating 177Lu-PSMA-617 Radioligand Therapy in Advanced Prostate Cancer Patients. J. Nucl. Med. 2017, 58, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Bronzel, M.; Apostolidis, C.; Weichert, W.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Targeted α-Therapy of Metastatic Castration-Resistant Prostate Cancer with 225Ac-PSMA-617: Dosimetry Estimate and Empiric Dose Finding. J. Nucl. Med. 2017, 58, 1624–1631. [Google Scholar] [CrossRef]
- Wang, F.; Li, Z.; Feng, X.; Yang, D.; Lin, M. Advances in PSMA-targeted therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2022, 25, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Huang, G.; Song, H.; Chen, Y.; Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Röhrich, M.; Winter, H.; et al. 68Ga-FAPI PET/CT: Tracer Uptake in 28 Different Kinds of Cancer. J. Nucl. Med. 2019, 60, 801–805. [Google Scholar] [CrossRef]
- Calais, J. FAP: The Next Billion Dollar Nuclear Theranostics Target? J. Nucl. Med. 2020, 61, 163–165. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of Quinoline-Based Theranostic Ligands for the Targeting of Fibroblast Activation Protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, J.; Pang, Y.; Fu, K.; Shang, Q.; Wu, H.; Sun, L.; Lin, Q.; Chen, H. Fibroblast activation protein-based theranostics in cancer research: A state-of-the-art review. Theranostics 2022, 12, 1557–1569. [Google Scholar] [CrossRef] [PubMed]
- Roustaei, H.; Kiamanesh, Z.; Askari, E.; Sadeghi, R.; Aryana, K.; Treglia, G. Could Fibroblast Activation Protein (FAP)-Specific Radioligands Be Considered as Pan-Tumor Agents? Contrast Media Mol. Imaging 2022, 2022, 3948873. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu, S.; Kovan, B.; Sanli, Y.; Buyukkaya, F.; Has Simsek, D.; Özkan, Z.G.; Isik, E.G.; Ekenel, M.; Turkmen, C. Safety of Fibroblast Activation Protein-Targeted Radionuclide Therapy by a Low-Dose Dosimetric Approach Using 177Lu-FAPI04. Clin. Nucl. Med. 2021, 46, 641–646. [Google Scholar] [CrossRef]
- Rathke, H.; Fuxius, S.; Giesel, F.L.; Lindner, T.; Debus, J.; Haberkorn, U.; Kratochwil, C. Two Tumors, One Target: Preliminary Experience With 90Y-FAPI Therapy in a Patient With Metastasized Breast and Colorectal Cancer. Clin. Nucl. Med. 2021, 46, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Assadi, M.; Rekabpour, S.J.; Jafari, E.; Divband, G.; Nikkholgh, B.; Amini, H.; Kamali, H.; Ebrahimi, S.; Shakibazad, N.; Jokar, N.; et al. Feasibility and Therapeutic Potential of 177Lu-Fibroblast Activation Protein Inhibitor-46 for Patients With Relapsed or Refractory Cancers: A Preliminary Study. Clin. Nucl. Med. 2021, 46, e523–e530. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Pang, Y.; Zhao, L.; Lin, L.; Wu, H.; Sun, L.; Lin, Q.; Chen, H. FAP-targeted radionuclide therapy with [177Lu]Lu-FAPI-46 in metastatic nasopharyngeal carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1767–1769. [Google Scholar] [CrossRef]
- Ferdinandus, J.; Fragoso Costa, P.; Kessler, L.; Weber, M.; Hirmas, N.; Kostbade, K.; Bauer, S.; Schuler, M.; Ahrens, M.; Schildhaus, H.U.; et al. Initial clinical experience with 90Y-FAPI-46 radioligand therapy for advanced stage solid tumors: A case series of nine patients. J. Nucl. Med. 2022, 63, 727–734. [Google Scholar] [CrossRef]
- Fu, H.; Huang, J.; Sun, L.; Wu, H.; Chen, H. FAP-Targeted Radionuclide Therapy of Advanced Radioiodine-Refractory Differentiated Thyroid Cancer With Multiple Cycles of 177 Lu-FAPI-46. Clin. Nucl. Med. 2022, 47, 906–907. [Google Scholar] [CrossRef]
- Fendler, W.P.; Pabst, K.M.; Kessler, L.; Fragoso Costa, P.; Ferdinandus, J.; Weber, M.; Lippert, M.; Lueckerath, K.; Umutlu, L.; Kostbade, K.; et al. Safety and Efficacy of 90Y-FAPI-46 Radioligand Therapy in Patients with Advanced Sarcoma and Other Cancer Entities. Clin. Cancer Res. 2022, 28, 4346–4353. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Rathke, H.; Fink, R.; Dendl, K.; Debus, J.; Mier, W.; Jäger, D.; Lindner, T.; Haberkorn, U. [153Sm]Samarium-labeled FAPI-46 radioligand therapy in a patient with lung metastases of a sarcoma. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3011–3013. [Google Scholar] [CrossRef]
- Zhang, P.; Xu, M.; Ding, J.; Chen, J.; Zhang, T.; Huo, L.; Liu, Z. Fatty acid-conjugated radiopharmaceuticals for fibroblast activation protein-targeted radiotherapy. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, P.; Ding, J.; Chen, J.; Huo, L.; Liu, Z. Albumin Binder-Conjugated Fibroblast Activation Protein Inhibitor Radiopharmaceuticals for Cancer Therapy. J. Nucl. Med. 2022, 63, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Xu, P.; Shi, M.; Liu, J.; Zeng, X.; Zhang, Y.; Shi, C.; Li, J.; Guo, Z.; Zhang, X.; et al. Evans blue-modified radiolabeled fibroblast activation protein inhibitor as long-acting cancer therapeutics. Theranostics 2022, 12, 422–433. [Google Scholar] [CrossRef]
- Moon, E.S.; Ballal, S.; Yadav, M.P.; Bal, C.; Van Rymenant, Y.; Stephan, S.; Bracke, A.; Van der Veken, P.; De Meester, I.; Roesch, F. Fibroblast Activation Protein (FAP) targeting homodimeric FAP inhibitor radiotheranostics: A step to improve tumor uptake and retention time. Am. J. Nucl. Med. Mol. Imaging 2021, 11, 476–491. [Google Scholar] [PubMed]
- Zhao, L.; Chen, J.; Pang, Y.; Fang, J.; Fu, K.; Meng, L.; Zhang, X.; Guo, Z.; Wu, H.; Sun, L.; et al. Development of Fibroblast Activation Protein Inhibitor-Based Dimeric Radiotracers with Improved Tumor Retention and Antitumor Efficacy. Mol. Pharm. 2022, 19, 3640–3651. [Google Scholar] [CrossRef] [PubMed]
- Ballal, S.; Yadav, M.P.; Moon, E.S.; Kramer, V.S.; Roesch, F.; Kumari, S.; Bal, C. First-In-Human Results on the Biodistribution, Pharmacokinetics, and Dosimetry of [177Lu]Lu-DOTA.SA.FAPi and [177Lu]Lu-DOTAGA.(SA.FAPi)2. Pharmaceuticals 2021, 14, 1212. [Google Scholar] [CrossRef]
- Ballal, S.; Yadav, M.P.; Moon, E.S.; Roesch, F.; Kumari, S.; Agarwal, S.; Tripathi, M.; Sahoo, R.K.; Mangu, B.S.; Tupalli, A.; et al. Novel Fibroblast Activation Protein Inhibitor-Based Targeted Theranostics for Radioiodine-Refractory Differentiated Thyroid Cancer Patients: A Pilot Study. Thyroid 2022, 32, 65–77. [Google Scholar] [CrossRef]
- Baum, R.P.; Schuchardt, C.; Singh, A.; Chantadisai, M.; Robiller, F.C.; Zhang, J.; Mueller, D.; Eismant, A.; Almaguel, F.; Zboralski, D.; et al. Feasibility, Biodistribution and Preliminary Dosimetry in Peptide-Targeted Radionuclide Therapy (PTRT) of Diverse Adenocarcinomas using 177Lu-FAP-2286: First-in-Human Results. J. Nucl. Med. 2022, 63, 415–423. [Google Scholar] [CrossRef]
- Millul, J.; Bassi, G.; Mock, J.; Elsayed, A.; Pellegrino, C.; Zana, A.; Dakhel Plaza, S.; Nadal, L.; Gloger, A.; Schmidt, E.; et al. An ultra-high-affinity small organic ligand of fibroblast activation protein for tumor-targeting applications. Proc. Natl. Acad. Sci. USA 2021, 118, e2101852118. [Google Scholar] [CrossRef]
- Bartoli, F.; Elsinga, P.; Nazario, L.R.; Zana, A.; Galbiati, A.; Millul, J.; Migliorini, F.; Cazzamalli, S.; Neri, D.; Slart, R.H.J.A.; et al. Automated Radiosynthesis, Preliminary In Vitro/In Vivo Characterization of OncoFAP-Based Radiopharmaceuticals for Cancer Imaging and Therapy. Pharmaceuticals 2022, 15, 958. [Google Scholar] [CrossRef]
- Galbiati, A.; Zana, A.; Bocci, M.; Millul, J.; Elsayed, A.; Mock, J.; Neri, D.; Cazzamalli, S. A Dimeric FAP-Targeting Small-Molecule Radioconjugate with High and Prolonged Tumor Uptake. J. Nucl. Med. 2022, 63, 1852–1858. [Google Scholar] [CrossRef]
- Liu, Y.; Watabe, T.; Kaneda-Nakashima, K.; Shirakami, Y.; Naka, S.; Ooe, K.; Toyoshima, A.; Nagata, K.; Haberkorn, U.; Kratochwil, C.; et al. Fibroblast activation protein targeted therapy using [177Lu]FAPI-46 compared with [225Ac]FAPI-46 in a pancreatic cancer model. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Altmann, A.; Krämer, S.; Kleist, C.; Loktev, A.; Kratochwil, C.; Giesel, F.; Mier, W.; Marme, F.; Debus, J.; et al. Design and Development of 99mTc-Labeled FAPI Tracers for SPECT Imaging and 188Re Therapy. J. Nucl. Med. 2020, 61, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Giesel, F.L.; Kratochwil, C.; Serfling, S.E. Radioligands Targeting Fibroblast Activation Protein (FAP). Cancers 2021, 13, 5744. [Google Scholar] [CrossRef] [PubMed]
- Imlimthan, S.; Moon, E.S.; Rathke, H.; Afshar-Oromieh, A.; Rösch, F.; Rominger, A.; Gourni, E. New Frontiers in Cancer Imaging and Therapy Based on Radiolabeled Fibroblast Activation Protein Inhibitors: A Rational Review and Current Progress. Pharmaceuticals 2021, 14, 1023. [Google Scholar] [CrossRef]
- Pazzaglia, S.; Pioli, C. Multifaceted role of PARP-1 in DNA repair and inflammation: Pathological and therapeutic implications in cancer and non-cancer diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef]
- Chan, C.Y.; Tan, K.V.; Cornelissen, B. PARP Inhibitors in Cancer Diagnosis and Therapy. Clin. Cancer Res. 2021, 27, 1585–1594. [Google Scholar] [CrossRef]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef]
- Carney, B.; Kossatz, S.; Reiner, T. Molecular Imaging of PARP. J. Nucl. Med. 2017, 58, 1025–1030. [Google Scholar] [CrossRef]
- Ambur Sankaranarayanan, R.; Kossatz, S.; Weber, W.; Beheshti, M.; Morgenroth, A.; Mottaghy, F.M. Advancements in PARP1 Targeted Nuclear Imaging and Theranostic Probes. J. Clin. Med. 2020, 9, 2130. [Google Scholar] [CrossRef]
- Jannetti, S.A.; Carlucci, G.; Carney, B.; Kossatz, S.; Shenker, L.; Carter, L.M.; Salinas, B.; Brand, C.; Sadique, A.; Donabedian, P.L.; et al. PARP-1-Targeted Radiotherapy in Mouse Models of Glioblastoma. J. Nucl. Med. 2018, 59, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Pacelli, A.; Nader, M.; Kossatz, S. DNA Repair Enzyme Poly(ADP-Ribose) Polymerase 1/2 (PARP1/2)-Targeted Nuclear Imaging and Radiotherapy. Cancers 2022, 14, 1129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, H.; Mpoy, C.; Afrin, S.; Rogers, B.E.; Garbow, J.R.; Katzenellenbogen, J.A.; Xu, J. Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as PARP-1-Targeting Agents. Biomedicines 2021, 9, 565. [Google Scholar] [CrossRef] [PubMed]
- Destro, G.; Chen, Z.; Chan, C.Y.; Fraser, C.; Dias, G.; Mosley, M.; Guibbal, F.; Gouverneur, V.; Cornelissen, B. A radioiodinated rucaparib analogue as an Auger electron emitter for cancer therapy. Nucl. Med. Biol. 2022, 116–117, 108312. [Google Scholar] [CrossRef] [PubMed]
- Makvandi, M.; Lee, H.; Puentes, L.N.; Reilly, S.W.; Rathi, K.S.; Weng, C.C.; Chan, H.S.; Hou, C.; Raman, P.; Martinez, D.; et al. Targeting PARP-1 with Alpha-Particles Is Potently Cytotoxic to Human Neuroblastoma in Preclinical Models. Mol. Cancer Ther. 2019, 18, 1195–1204. [Google Scholar] [CrossRef]
- Lee, H.; Riad, A.; Martorano, P.; Mansfield, A.; Samanta, M.; Batra, V.; Mach, R.H.; Maris, J.M.; Pryma, D.A.; Makvandi, M. PARP-1-Targeted Auger Emitters Display High-LET Cytotoxic Properties In Vitro but Show Limited Therapeutic Utility in Solid Tumor Models of Human Neuroblastoma. J. Nucl. Med. 2020, 61, 850–856. [Google Scholar] [CrossRef]
- Pirovano, G.; Jannetti, S.A.; Carter, L.M.; Sadique, A.; Kossatz, S.; Guru, N.; Demétrio De Souza França, P.; Maeda, M.; Zeglis, B.M.; Lewis, J.S.; et al. Targeted Brain Tumor Radiotherapy Using an Auger Emitter. Clin. Cancer Res. 2020, 26, 2871–2881. [Google Scholar] [CrossRef]
- Ambur Sankaranarayana, R.; Florea, A.; Allekotte, S.; Vogg, A.T.J.; Maurer, J.; Schäfer, L.; Bolm, C.; Terhorst, S.; Classen, A.; Bauwens, M.; et al. PARP targeted Auger emitter therapy with [125I]PARPi-01 for triple-negative breast cancer. EJNMMI Res. 2022, 12, 60. [Google Scholar] [CrossRef]
- Brown, J.M. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007, 435, 297–321. [Google Scholar]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic Anhydrase IX: Regulation and role in cancer. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2014; pp. 199–219. [Google Scholar]
- Singh, S.; Lomelino, C.L.; Mboge, M.Y.; Frost, S.C.; McKenna, R. Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Lin, K.S.; Bénard, F. Past, Present, and Future: Development of Theranostic Agents Targeting Carbonic Anhydrase IX. Theranostics 2017, 7, 4322–4339. [Google Scholar] [CrossRef] [PubMed]
- Iikuni, S.; Ono, M.; Watanabe, H.; Shimizu, Y.; Sano, K.; Saji, H. Cancer radiotheranostics targeting carbonic anhydrase-IX with 111In- and 90Y-labeled ureidosulfonamide scaffold for SPECT imaging and radionuclide-based therapy. Theranostics 2018, 8, 2992–3006. [Google Scholar] [CrossRef]
- Janoniene, A.; Petrikaite, V. In Search of Advanced Tumor Diagnostics and Treatment: Achievements and Perspectives of Carbonic Anhydrase IX Targeted Delivery. Mol. Pharm. 2020, 17, 1800–1815. [Google Scholar] [CrossRef] [PubMed]
- Shahrokhi, P.; Farahani, A.M.; Tamaddondar, M. Radiolabeled vitamins as the potential diagnostic probes for targeted tumor imaging. Bioorg. Chem. 2022, 122, 105717. [Google Scholar] [CrossRef]
- Jurczyk, M.; Jelonek, K.; Musiał-Kulik, M.; Beberok, A.; Wrześniok, D.; Kasperczyk, J. Single- versus Dual-Targeted Nanoparticles with Folic Acid and Biotin for Anticancer Drug Delivery. Pharmaceutics 2021, 13, 326. [Google Scholar] [CrossRef]
- Jallinoja, V.I.J.; Houghton, J.L. Current Landscape in Clinical Pretargeted Radioimmunoimaging and Therapy. J. Nucl. Med. 2021, 62, 1200–1206. [Google Scholar] [CrossRef]
- Russell-Jones, G.; McTavish, K.; McEwan, J.; Rice, J.; Nowotnik, D. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J. Inorg. Biochem. 2004, 98, 1625–1633. [Google Scholar] [CrossRef]
- Low, P.S.; Kularatne, S.A. Folate-targeted therapeutic and imaging agents for cancer. Curr. Opin. Chem. Biol. 2009, 13, 256–262. [Google Scholar] [CrossRef]
- Xia, W.; Low, P.S. Folate-targeted therapies for cancer. J. Med. Chem. 2010, 53, 6811–6824. [Google Scholar] [CrossRef]
- Muller, C. Folate based radiopharmaceuticals for imaging and therapy of cancer and inflammation. Curr. Pharm. Des. 2012, 1, 1058–1083. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Guzik, P.; Siwowska, K.; Cohrs, S.; Schmid, R.M.; Schibli, R. Combining Albumin-Binding Properties and Interaction with Pemetrexed to Improve the Tissue Distribution of Radiofolates. Molecules 2018, 23, 1465. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Guzik, P.; Deberle, L.M.; Busslinger, S.D.; Landolt, T.; Schibli, R.; Müller, C. Design and Evaluation of Novel Albumin-Binding Folate Radioconjugates: Systematic Approach of Varying the Linker Entities. Mol. Pharm. 2022, 19, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Boss, S.D.; Ametamey, S.M. Development of Folate Receptor-Targeted PET Radiopharmaceuticals for Tumor Imaging-A Bench-to-Bedside Journey. Cancers 2020, 12, 1508. [Google Scholar] [CrossRef]
- Müller, C.; Schibli, R. Prospects in folate receptor-targeted radionuclide therapy. Front. Oncol. 2013, 3, 249. [Google Scholar] [CrossRef]
- Kuda-Wedagedara, A.N.W.; Workinger, J.L.; Nexo, E.; Doyle, R.P.; Viola-Villegas, N. 89Zr-Cobalamin PET Tracer: Synthesis, Cellular Uptake, and Use for Tumor Imaging. ACS Omega 2017, 2, 6314–6320. [Google Scholar] [CrossRef]
- Gendron, L.N.; Zites, D.C.; LaRochelle, E.P.M.; Gunn, J.R.; Pogue, B.W.; Shell, T.A.; Shell, J.R. Tumor targeting vitamin B12 derivatives for X-ray induced treatment of pancreatic adenocarcinoma. Photodiagnosis Photodyn. Ther. 2020, 30, 101637. [Google Scholar] [CrossRef]
- Snyder, F.; Wood, R. Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues. Cancer Res. 1969, 29, 251–257. [Google Scholar]
- Vink, S.R.; van Blitterswijk, W.J.; Schellens, J.H.; Verheij, M. Rationale and clinical application of alkylphospholipid analogues in combination with radiotherapy. Cancer Treat. Rev. 2007, 33, 191–202. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts and clusters of apoptotic signaling molecule-enriched rafts in cancer therapy. Future Oncol. 2010, 6, 811–821. [Google Scholar] [CrossRef]
- Weichert, J.P.; Clark, P.A.; Kandela, I.K.; Vaccaro, A.M.; Clarke, W.; Longino, M.A.; Pinchuk, A.N.; Farhoud, M.; Swanson, K.I.; Floberg, J.M.; et al. Alkylphosphocholine analogs for broad-spectrum cancer imaging and therapy. Sci. Transl. Med. 2014, 6, 240ra75. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.L.; Schwendner, S.W.; Counsell, R.E. Potential tumor or organ-imaging agents. 30. Radioiodinated phospholipid ethers. J. Med. Chem. 1989, 32, 2142–2147. [Google Scholar] [CrossRef] [PubMed]
- Plotzke, K.P.; Haradahira, T.; Stancato, L.; Olken, N.M.; Skinner, S.; Gross, M.D.; Wahl, R.L.; Counsell, R.E. Selective localization of radioiodinated alkylphosphocholine derivatives in tumors. Int. J. Rad. Appl. Instrum. B 1992, 19, 765–773. [Google Scholar] [CrossRef]
- Rampy, M.A.; Chou, T.S.; Pinchuk, A.N.; Skinner, R.W.; Gross, M.D.; Fisher, S.; Wahl, R.; Counsell, R.E. Synthesis and biological evaluation of radioiodinated phospholipid ether analogs. Nucl. Med. Biol. 1995, 22, 505–512. [Google Scholar] [CrossRef]
- Grudzinski, J.J.; Hall, L.T.; Cho, S.; Liu, G.; Traynor, A.; Lee, M.H.; Longino, M.; Pinchuk, A.; Jaskowiak, C.; Bednarz, B.; et al. Clinical Imaging and Dosimetry of a Pan-Cancer Targeting Alkylphosphocholine Analog, [124I]I-NM404. Radiation 2022, 2, 215–227. [Google Scholar] [CrossRef]
- Pinchuk, A.N.; Rampy, M.A.; Longino, M.A.; Skinner, R.W.; Gross, M.D.; Weichert, J.P.; Counsell, R.E. Synthesis and structure-activity relationship effects on the tumor avidity of radioiodinated phospholipid ether analogues. J. Med. Chem. 2006, 49, 2155–2165. [Google Scholar] [CrossRef]
- Morris, Z.S.; Weichert, J.P.; Saker, J.; Armstrong, E.A.; Besemer, A.; Bednarz, B.; Kimple, R.J.; Harari, P.M. Therapeutic combination of radiolabeled CLR1404 with external beam radiation in head and neck cancer model systems. Radiother. Oncol. 2015, 116, 504–509. [Google Scholar] [CrossRef]
- Lubner, S.J.; Mullvain, J.; Perlman, S.; Pishvaian, M.; Mortimer, J.; Oliver, K.; Heideman, J.; Hall, L.; Weichert, J.; Liu, G. A Phase 1, Multi-Center, Open-Label, Dose-Escalation Study of 131I-CLR1404 in Subjects with Relapsed or Refractory Advanced Solid Malignancies. Cancer Investig. 2015, 33, 483–489. [Google Scholar] [CrossRef]
- Hall, L.T.; Titz, B.; Robins, H.I.; Bednarz, B.P.; Perlman, S.B.; Weichert, J.P.; Kuo, J.S. PET/CT imaging of the diapeutic alkylphosphocholine analog 124I-CLR1404 in high and low-grade brain tumors. Am. J. Nucl. Med. Mol. Imaging 2017, 7, 157–166. [Google Scholar]
- Baiu, D.C.; Marsh, I.R.; Boruch, A.E.; Shahi, A.; Bhattacharya, S.; Jeffery, J.J.; Zhao, Q.; Hall, L.T.; Weichert, J.P.; Bednarz, B.P.; et al. Targeted Molecular Radiotherapy of Pediatric Solid Tumors Using a Radioiodinated Alkyl-Phospholipid Ether Analog. J. Nucl. Med. 2018, 59, 244–250. [Google Scholar] [CrossRef]
- Hall, L.T.; Titz, B.; Baidya, N.; van der Kolk, A.G.; Robins, H.I.; Otto, M.; Perlman, S.B.; Weichert, J.P.; Kuo, J.S. [124I]CLR1404 PET/CT in High-Grade Primary and Metastatic Brain Tumors. Mol. Imaging Biol. 2020, 22, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Shahi, A.; Weiss, G.E.; Bhattacharya, S.; Baiu, D.C.; Marino, R.; Pula, T.; Callander, N.S.; Asimakopoulos, F.; Otto, M. Targeted treatment of multiple myeloma with a radioiodinated small molecule radiopharmaceutical. Leuk. Lymphoma 2021, 62, 1518–1521. [Google Scholar] [CrossRef] [PubMed]
- Longcor, J.; Callander, N.; Oliver, K.; Chanan-Khan, A.; Ailawadhi, S. Iopofosine I-131 treatment in late-line patients with relapsed/refractory multiple myeloma post anti-BCMA immunotherapy. Blood Cancer J. 2022, 12, 130. [Google Scholar] [CrossRef]
- Grudzinski, J.J.; Hernandez, R.; Marsh, I.; Patel, R.B.; Aluicio-Sarduy, E.; Engle, J.; Morris, Z.; Bednarz, B.; Weichert, J. Preclinical Characterization of 86/90Y-NM600 in a Variety of Murine and Human Cancer Tumor Models. J. Nucl. Med. 2019, 60, 1622–1628. [Google Scholar] [CrossRef]
- Hernandez, R.; Grudzinski, J.J.; Aluicio-Sarduy, E.; Massey, C.F.; Pinchuk, A.N.; Bitton, A.N.; Patel, R.; Zhang, R.; Rao, A.V.; Iyer, G.; et al. 177Lu-NM600 Targeted Radionuclide Therapy Extends Survival in Syngeneic Murine Models of Triple-Negative Breast Cancer. J. Nucl. Med. 2020, 61, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Norain, A.; Dadachova, E. Targeted Radionuclide Therapy of Melanoma. Semin. Nucl. Med. 2016, 46, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.J.H.; Malo, M.E.; Jiao, R.; Dadachova, E. Targeting Melanin in Melanoma with Radionuclide Therapy. Int. J. Mol. Sci. 2022, 23, 9520. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.E.; Kricker, A.; Waxweiler, W.T.; Dillon, P.M.; Busman, K.J.; From, L.; Groben, P.A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; et al. Comparison of clinicopathologic features and survival of histopathologically amelanotic and pigmented melanomas: A population-based study. JAMA Dermatol. 2014, 150, 1306–1314. [Google Scholar] [CrossRef]
- Klein, M.; Lotem, M.; Peretz, T.; Zwas, S.T.; Mizrachi, S.; Liberman, Y.; Chisin, R.; Schachter, J.; Ron, I.G.; Iosilevsky, G.; et al. Safety and efficacy of 188-rhenium-labeled antibody to melanin in patients with metastatic melanoma. J. Skin Cancer 2013, 2013, 828329. [Google Scholar] [CrossRef]
- Rouanet, J.; Quintana, M.; Auzeloux, P.; Cachin, F.; Degoul, F. Benzamide derivative radiotracers targeting melanin for melanoma imaging and therapy: Preclinical/clinical development and combination with other treatments. Pharmacol. Ther. 2021, 224, 107829. [Google Scholar] [CrossRef]
- Mohammed, A.; Nicholl, C.; Titsch, U.; Eisenhut, M. Radioiodinated N-(alkylaminoalkyl)-substituted 4-methoxy-, 4-hydroxy-, and 4-aminobenzamides: Biological investigations for the improvement of melanoma-imaging agents. Nucl. Med. Biol. 1997, 24, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Eisenhut, M.; Hull, W.E.; Mohammed, A.; Mier, W.; Lay, D.; Just, W.; Gorgas, K.; Lehmann, W.D.; Haberkorn, U. Radioiodinated N-(2-diethylaminoethyl)benzamide derivatives with high melanoma uptake: Structure-affinity relationships, metabolic fate, and intracellular localization. J. Med. Chem. 2000, 43, 3913–3922. [Google Scholar] [CrossRef] [PubMed]
- Auzeloux, P.; Papon, J.; Pasqualini, R.; Madelmont, J.C. Synthesis and biodistribution of a new oxo-technetium-99m bis(aminothiol) complex as a potential melanoma tracer. J. Med. Chem. 2001, 44, 1116–1121. [Google Scholar] [CrossRef]
- Oltmanns, D.; Eisenhut, M.; Mier, W.; Haberkorn, U. Benzamides as melanotropic carriers for radioisotopes, metals, cytotoxic agents and as enzyme inhibitors. Curr. Med. Chem. 2009, 16, 2086–2094. [Google Scholar] [CrossRef]
- Michelot, J.M.; Moreau, M.F.; Veyre, A.J.; Bonafous, J.F.; Bacin, F.J.; Madelmont, J.C.; Bussiere, F.; Souteyrand, P.A.; Mauclaire, L.P.; Chossat, F.M.; et al. Phase II scintigraphic clinical trial of malignant melanoma and metastases with iodine-123-N-(2-diethylaminoethyl 4-iodobenzamide). J. Nucl. Med. 1993, 34, 1260–1266. [Google Scholar] [PubMed]
- Link, E.M. Targeting melanoma with 211At/131I-methylene blue: Preclinical and clinical experience. Hybridoma 1999, 18, 77–82. [Google Scholar] [CrossRef]
- Joyal, J.L.; Barrett, J.A.; Marquis, J.C.; Chen, J.; Hillier, S.M.; Maresca, K.P.; Boyd, M.; Gage, K.; Nimmagadda, S.; Kronauge, J.F.; et al. Preclinical evaluation of an 131I-labeled benzamide for targeted radiotherapy of metastatic melanoma. Cancer Res. 2010, 70, 4045–4053. [Google Scholar] [CrossRef]
- Degoul, F.; Borel, M.; Jacquemot, N.; Besse, S.; Communal, Y.; Mishellany, F.; Papon, J.; Penault-Llorca, F.; Donnarieix, D.; Doly, M.; et al. In vivo efficacy of melanoma internal radionuclide therapy with a 131I-labelled melanin-targeting heteroarylcarboxamide molecule. Int. J. Cancer 2013, 133, 1042–1053. [Google Scholar] [CrossRef]
- Xu, X.; Yuan, L.; Gai, Y.; Liu, Q.; Yin, L.; Jiang, Y.; Wang, Y.; Zhang, Y.; Lan, X. Targeted radiotherapy of pigmented melanoma with 131I-5-IPN. J. Exp. Clin. Cancer Res. 2018, 37, 306. [Google Scholar] [CrossRef]
- Mier, W.; Kratochwil, C.; Hassel, J.C.; Giesel, F.L.; Beijer, B.; Babich, J.W.; Friebe, M.; Eisenhut, M.; Enk, A.; Haberkorn, U. Radiopharmaceutical therapy of patients with metastasized melanoma with the melanin-binding benzamide 131I-BA52. J. Nucl. Med. 2014, 55, 9–14. [Google Scholar] [CrossRef]
- Thivat, E.; Rouanet, J.; Auzeloux, P.; Sas, N.; Jouberton, E.; Levesque, S.; Billoux, T.; Mansard, S.; Molnar, I.; Chanchou, M.; et al. Phase I study of [131I] ICF01012, a targeted radionuclide therapy, in metastatic melanoma: MELRIV-1 protocol. BMC Cancer 2022, 22, 417. [Google Scholar] [CrossRef] [PubMed]
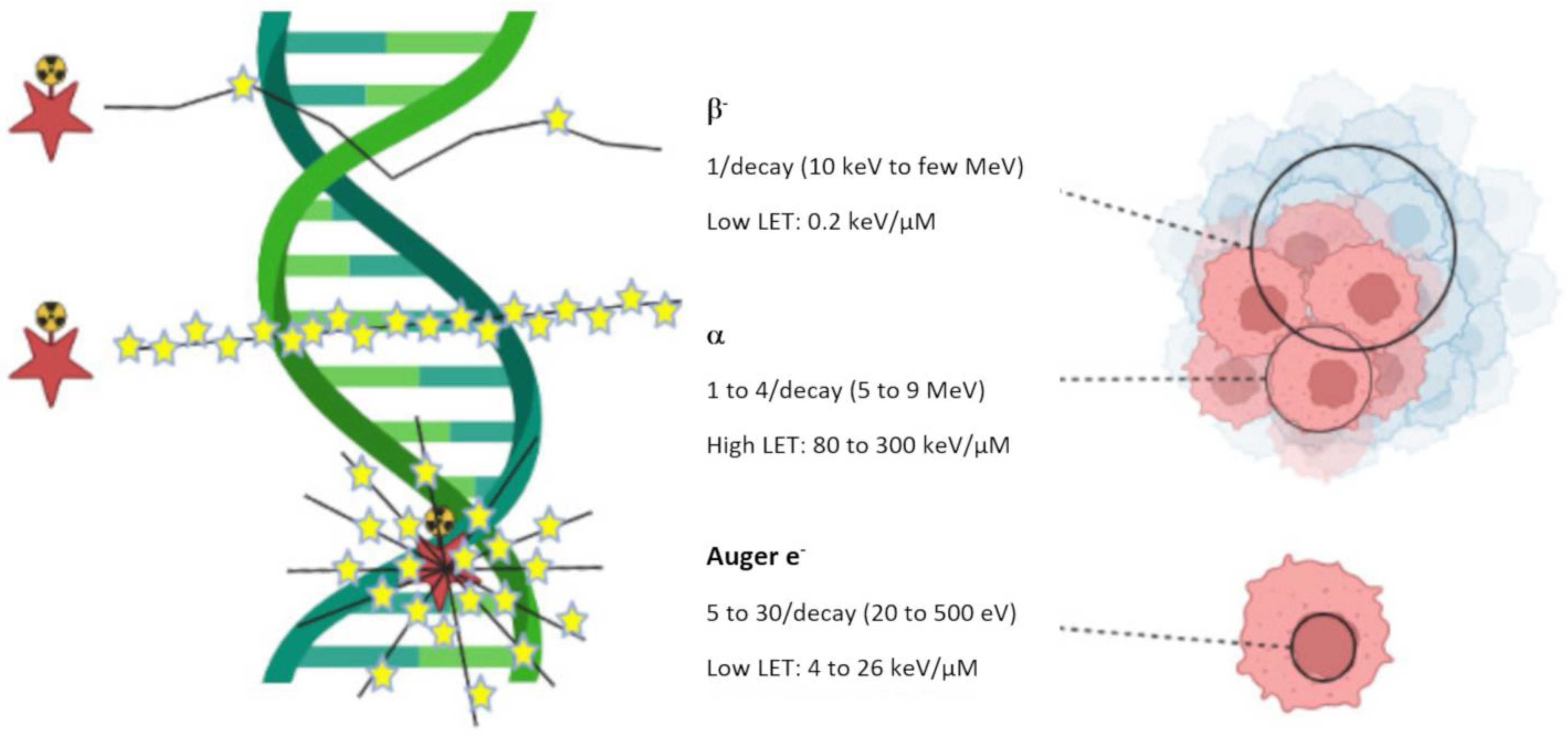
 ) represent disintegrations.
) represent disintegrations.
) represent disintegrations.
) represent disintegrations.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radionuclide | Half-life | Energy (MeV) | Eγ (keV) | Tissue Penetration Range (mm) |
|---|---|---|---|---|
| β-emitter | ||||
| 90Y | 2.7 days | 2.284 | / | 12 |
| 131I | 8 days | 0.81 | 0.364 | 2.4 |
| 161Tb | 6.9 days | 0.593 | 74.6 | 3 |
| 177Lu | 6.7 days | 0.497 | 208113 | 2.2 |
| 188Re | 17 h | 2.118 | 155 | 11 |
| α-emitter | ||||
| 149Tb | 4.1 h | 3.97 | Multiple emissions (165–800) | <100 µm |
| 211At | 7.2 h | 7.45 | 85 (X-ray) | |
| 212Pb/212Bi * | 10.6 h | 8.78 | 238, 300 | |
| 213Bi | 0.8 h | 8.38 | 440 | |
| 223Ra | 11.4 days | 5.71, 6.82, 7.39, 6.62 | 270 | |
| 225Ac | 10 days | 5.8, 6.3, 7.1, 8.38 | 218, 440 (from daughters) | |
| 227Th | 18.7 days | 6.14, 5.71, 6.82, 7.39, 6.62 | 236 | |
| Auger e− emitter | ||||
| 111In | 2.8 days | 0.007 | 405 | <1 µm |
| 125I | 60 days | 0.019 | 42 |
| Peptide | Receptor | Tumor Expression |
|---|---|---|
| α-Melanocyte-stimulating hormone | MCR1, MCR3, and MCR5 | Melanomas |
| Bombesin/Gastrin-releasing peptide | BB1, BB2 (GRPR), BB3, and BB4 | Glioblastomas, prostate, breast, pancreatic, gastric, colorectal cancers, and small cell lung |
| Cholecystokinin/gastrin | CCK1, and CCK2 | Adenomas, astrocytomas, gastrointestinal and ovarian stromal tumors, medullary thyroid, pancreatic and small cell lung cancers |
| Epidermal Growth Factor | EGFR | Breast cancer |
| Exendin | GLP-1 | Gastrinomas, insulinomas, medullary thyroid carcinomas, paragangliomas and pheochromocytomas |
| Gonadotropin-releasing hormone | GnRH-R | Breast and prostate cancers |
| Neuropeptide Y | Y1, Y2, Y4, and Y5 | Breast, ovary, adrenal, brain, kidney, GI-tract, and bone (Ewing’s sarcoma) |
| Neurotensin | NTR1, NTR2, and NTR3 | Breast, colon, pancreatic, prostate, small cell lung cancers, and meningiomas |
| RGD | αVβ3 integrin | Tumor-induced angiogenesis |
| SDF-1α/CXCL12 | CXCR4, and CXCR7 | Leukemias, lymphomas, melanomas, brain, breast, kidney, lung, ovarian, pancreas, and prostate tumors |
| Somatostatin | Sstr1, sstr2, sstr3, sstr4, and sstr5 | Neuroendocrine tumors, lymphomas, paragangliomas, brain, breast, renal, and small cell lung cancers |
| Substance P | NK1, NK2, and NK3 | Glial tumors, breast, medullary thyroid, pancreas, and small cell lung cancers |
| Vasoactive intestinal peptide | VPAC1, and VPAC2 | Bladder, breast, gastrointestinal, non-small cell lung, ovarian, pancreatic, and prostate cancers |
| Peptide | Peptidic Sequence |
|---|---|
| OC Octreotide | d-Phe-cyclo(Cys-Phe-d-Trp-Lys-Thr-Cys)Thr(ol) |
| LAN Lanreotide | β-d-Nal-cyclo(Cys-Tyr-d-Trp-Lys-Val-Cys)Thr-NH2 |
| VAP Vapreotide | d-Phe-cyclo(Cys-Phe-d-Trp-Lys-Val-Cys)Trp-NH2 |
| TOC [Tyr3]-Octreotide | d-Phe-cyclo(Cys-Tyr-d-Trp-Lys-Thr-Cys)Thr(ol) |
| TATE [Tyr3]-Octreotate | d-Phe-cyclo(Cys-Tyr-d-Trp-Lys-Thr-Cys)Thr |
| NOC [1-Nal3]-Octreotide | d-Phe-cyclo(Cys-1-Nal-d-Trp-Lys-Thr-Cys)Thr(ol) |
| SOM230 Pasireotide | Cyclo(Hyp(Unk)-Phg- d-Trp-Lys-Tyr(Bn)-Phe) |
| P2045 Tozaride | Ser-Thr-Cys(Trt)-Phe(4-NH2)-(β-DAP)-CH2CO-S-cyclo((N-Me)HCy-Phe-Tyr-d-Trp-Lys-Thr) |
| Target | Name | Structure |
|---|---|---|
| SSTR | DOTA-BASS |  |
| DOTA-LM3 |  | |
| DOTA-JR11 (Satoreotide) |  | |
| GRPR | DOTA-RM2 |  |
| NeoB |  | |
| NTR | 3BP-227 |  |
| Radionuclide | Agent |
|---|---|
| Approved Agents for Clinical Use | |
| Strontium-89 (β−) (50.5 d) | [89Sr]SrCl2—Metastron® |
| Samarium-153 (β−) (1.9 d) | EDTMP—Quadramet® |
| Rhenium-186 (β−) (3.7 d) | HEDP |
| Radium-223 (α) (11.4 d) | [223Ra]RaCl2—Xofigo® |
| Agents in Clinical Trials | |
| Rhenium-188 (β−) (17 h) | HEDP Zoledronic acid |
| Lutetium-177 (β−) (6.8 d) | EDTMP DOTMP * Zoledronic acid (Dotazol) |
| Holmium-166 (β−) (1.1 d) | DOTMP * |
| Tin-117m (CE) (13.6 d) | DTPA ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lepareur, N.; Ramée, B.; Mougin-Degraef, M.; Bourgeois, M. Clinical Advances and Perspectives in Targeted Radionuclide Therapy. Pharmaceutics 2023, 15, 1733. https://doi.org/10.3390/pharmaceutics15061733
Lepareur N, Ramée B, Mougin-Degraef M, Bourgeois M. Clinical Advances and Perspectives in Targeted Radionuclide Therapy. Pharmaceutics. 2023; 15(6):1733. https://doi.org/10.3390/pharmaceutics15061733
Chicago/Turabian StyleLepareur, Nicolas, Barthélémy Ramée, Marie Mougin-Degraef, and Mickaël Bourgeois. 2023. "Clinical Advances and Perspectives in Targeted Radionuclide Therapy" Pharmaceutics 15, no. 6: 1733. https://doi.org/10.3390/pharmaceutics15061733
APA StyleLepareur, N., Ramée, B., Mougin-Degraef, M., & Bourgeois, M. (2023). Clinical Advances and Perspectives in Targeted Radionuclide Therapy. Pharmaceutics, 15(6), 1733. https://doi.org/10.3390/pharmaceutics15061733