
Rewiring Drug Research and Development through Human Data-Driven Discovery (HD3)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
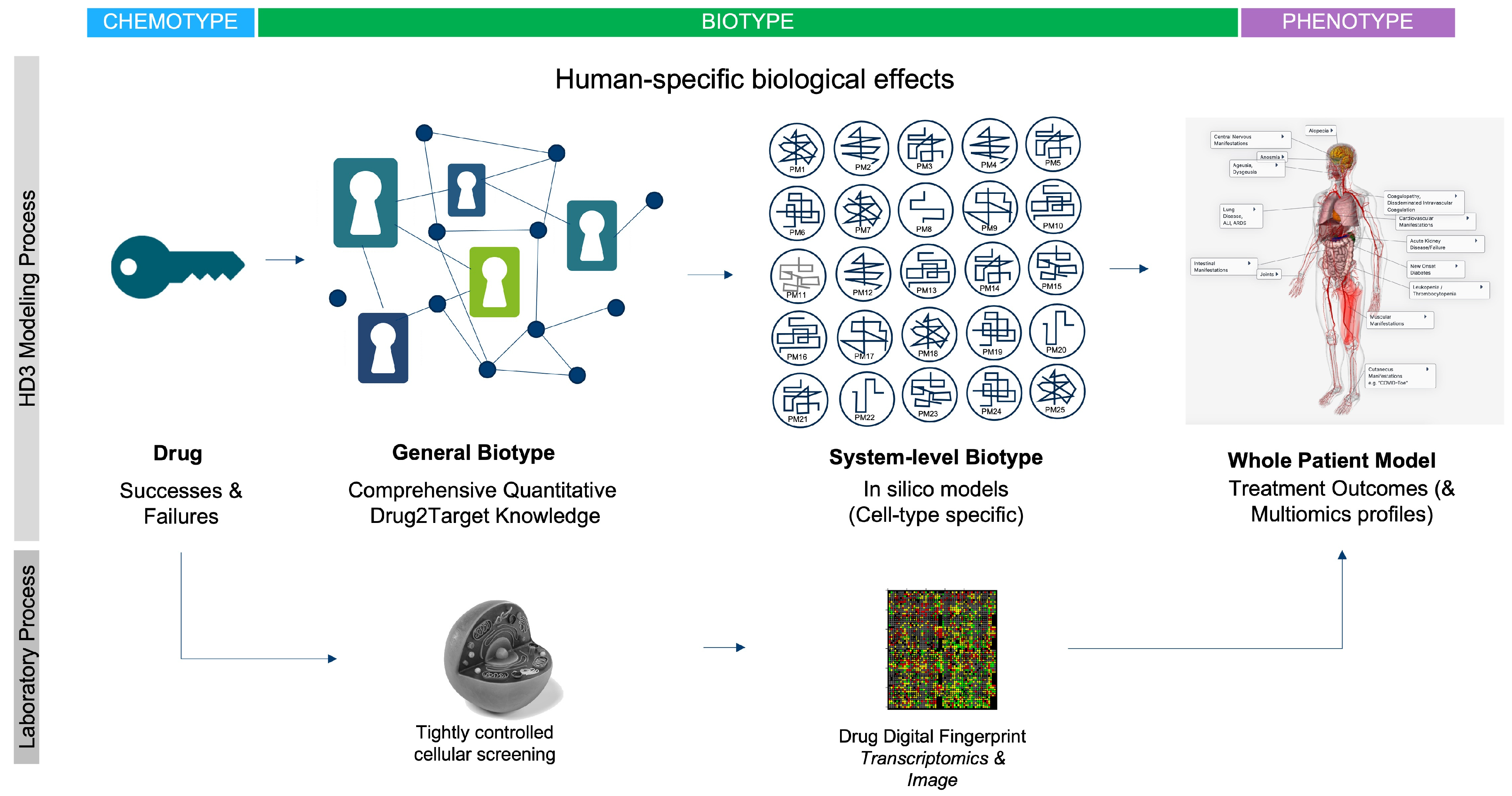
1. Introduction
2. Eroom’s Law and the Innovation Crisis
2.1. The Traditional Drug Discovery Paradigm
2.2. Industry-Level Challenges
2.2.1. Regulatory Oversight
2.2.2. The Drug “Innovation Chasm”
2.2.3. Mergers and Acquisitions
2.3. Science and Technology Challenges
2.3.1. Target-Based Discovery
2.3.2. Drug Promiscuity
2.3.3. The Reproducibility Crisis
2.3.4. The Problem with Model Systems
3. The “First Principles” Case for a Human Data-Driven Discovery (HD3) Paradigm
Human Data as a Driver for Systems-Based Discovery
4. Current Applications of the HD3 Approach
4.1. Application to the Analysis and Prediction of Adverse Events
Examples from the FDA’s Division of Applied Regulatory Science
4.2. Application of HD3 to Drug Repositioning and Combinatorial Therapy Design
5. Synopsis and Future Directions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchinson, L.; Kirk, R. High drug attrition rates—Where are we going wrong? Nat. Rev. Clin. Oncol. 2011, 8, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Barton, P.; Riley, R.J. A new paradigm for navigating compound property related drug attrition. Drug Discov. Today 2016, 21, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Evaluate Ltd. Evaluate Vantage 2020 Preview. Available online: https://www.evaluate.com/thought-leadership/vantage/evaluate-vantage-2020-preview#download (accessed on 15 January 2023).
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef]
- Davis, R.L. Mechanism of Action and Target Identification: A Matter of Timing in Drug Discovery. iScience 2020, 23, 101487. [Google Scholar] [CrossRef]
- Wyatt, P.; Gilbert, I.; Read, K.D.; Fairlamb, A.H. Target Validation: Linking Target and Chemical Properties to Desired Product Profile. Curr. Top. Med. Chem. 2011, 11, 1275–1283. [Google Scholar] [CrossRef]
- Morra, G.; Genoni, A.; Neves, M.; Merz, K.M., Jr.; Colombo, G. Molecular Recognition and Drug-Lead Identification: What Can Molecular Simulations Tell Us? Curr. Med. Chem. 2010, 17, 25–41. [Google Scholar] [CrossRef]
- Wang, S.; Dong, G.; Sheng, C. Structural simplification: An efficient strategy in lead optimization. Acta Pharm. Sin. B 2019, 9, 880–901. [Google Scholar] [CrossRef]
- Haley, B.; Roudnicky, F. Functional Genomics for Cancer Drug Target Discovery. Cancer Cell 2020, 38, 31–43. [Google Scholar] [CrossRef]
- Williams, M. Editorial overview: From Vioxx to Luckenbach: Drug discovery at a crossroads. Curr. Opin. Investig. Drugs 2005, 6, 17–20. [Google Scholar] [PubMed]
- Nissen, S. Rosiglitazone: A disappointing DREAM. Future Cardiol. 2007, 3, 491–492. [Google Scholar] [CrossRef] [PubMed]
- I Kaitin, K.; A DiMasi, J. Pharmaceutical Innovation in the 21st Century: New Drug Approvals in the First Decade, 2000–2009. Clin. Pharmacol. Ther. 2011, 89, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. Industrial Policy and International Competitiveness in the Pharmaceutical Industry. In Competitive Strategies in the Pharmaceutical Industry; Helms, R., Ed.; The American Enterprise Institute: Washington, DC, USA, 1996; pp. 107–129. [Google Scholar]
- Tufts Center Report on Trial Timelines. Available online: https://www.centerwatch.com/articles/25033-trend-of-longer-trial-timelines-is-likely-to-continue (accessed on 19 November 2022).
- LaMattina, J.L. The impact of mergers on pharmaceutical R&D. Nat. Rev. Drug Discov. 2011, 10, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Szabo, M.; Akusjärvi, S.S.; Saxena, A.; Liu, J.; Janebjer, G.C.; Kitambi, S.S. Cell and small animal models for phenotypic drug discovery. Drug Des. Dev. Ther. 2017, 11, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. [Google Scholar] [CrossRef]
- Luo, J. CRISPR/Cas9: From Genome Engineering to Cancer Drug Discovery. Trends Cancer 2016, 2, 313–324. [Google Scholar] [CrossRef]
- Bon, M.; Bilsland, A.; Bower, J.; McAulay, K. Fragment-based drug discovery—The importance of high-quality molecule libraries. Mol. Oncol. 2022, 16, 3761–3777. [Google Scholar] [CrossRef]
- Tewkesbury, D.H.; Robey, R.C.; Barry, P.J. Progress in precision medicine in cystic fibrosis: A focus on CFTR modulator therapy. Breathe 2021, 17, 210112. [Google Scholar] [CrossRef]
- Carofiglio, F.; Lopalco, A.; Lopedota, A.; Cutrignelli, A.; Nicolotti, O.; Denora, N.; Stefanachi, A.; Leonetti, F. Bcr-Abl Tyrosine Kinase Inhibitors in the Treatment of Pediatric CML. Int. J. Mol. Sci. 2020, 21, 4469. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Mestres, J.; Gregori-Puigjané, E.; Valverde, S.; Solé, R.V. Data completeness—The Achilles heel of drug-target networks. Nat. Biotechnol. 2008, 26, 983–984. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Peers, I.S.; Ceuppens, P.R.; Harbron, C. In search of preclinical robustness. Nat. Rev. Drug Discov. 2012, 11, 733–734. [Google Scholar] [CrossRef] [PubMed]
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 712. [Google Scholar] [CrossRef]
- Hackam, D.G.; Redelmeier, D.A. Translation of Research Evidence from Animals to Humans. JAMA 2006, 296, 1731–1732. [Google Scholar] [CrossRef] [PubMed]
- Kaste, M. Use of Animal Models Has Not Contributed to Development of Acute Stroke Therapies: Pro. Stroke 2005, 36, 2323–2324. [Google Scholar] [CrossRef]
- Horrobin, D.F. Modern biomedical research: An internally self-consistent universe with little contact with medical reality? Nat. Rev. Drug Discov. 2003, 2, 151–154. [Google Scholar] [CrossRef]
- First Principles Thinking. Available online: https://www.csc.edu/media/website/content-assets/documents/pdf/tlpec/First-Principles-Thinking.pdf (accessed on 12 December 2022).
- Workman, P.; Al-Lazikani, B.; A Clarke, P. Genome-based cancer therapeutics: Targets, kinase drug resistance and future strategies for precision oncology. Curr. Opin. Pharmacol. 2013, 13, 486–496. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- FDA Adverse Events Reporting System (FAERS) Public Dashboard. Available online: https://fis.fda.gov/sense/app/d10be6bb-494e-4cd2-82e4-0135608ddc13/sheet/7a47a261-d58b-4203-a8aa-6d3021737452/state/analysis (accessed on 13 January 2023).
- Sentinel Initiative. Available online: https://www.sentinelinitiative.org/ (accessed on 12 January 2023).
- Ball, R.; Robb, M.; Anderson, S.; Pan, G.D. The FDA’s sentinel initiative-A comprehensive approach to medical product surveillance. Clin. Pharmacol. Ther. 2016, 99, 265–268. [Google Scholar] [CrossRef] [PubMed]
- European Database of Suspected Adverse Drug Reaction Reports. Available online: https://www.adrreports.eu/en/index.html (accessed on 20 December 2022).
- UMC|VigiBase. Available online: https://www.who-umc.org/vigibase/vigibase/ (accessed on 28 December 2022).
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2014, 43, D789–D798. [Google Scholar] [CrossRef] [PubMed]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.; Fuchsberger, C.; Köttgen, A.; O’seaghdha, C.M.; Pattaro, C.; de Andrade, M.; Chasman, D.I.; Teumer, A.; Endlich, K.; Olden, M.; et al. Common Variants in Mendelian Kidney Disease Genes and Their Association with Renal Function. J. Am. Soc. Nephrol. 2013, 24, 2105–2117. [Google Scholar] [CrossRef]
- Pattaro, C.; Köttgen, A.; Teumer, A.; Garnaas, M.; Böger, C.A.; Fuchsberger, C.; Olden, M.; Chen, M.-H.; Tin, A.; Taliun, D.; et al. Genome-Wide Association and Functional Follow-Up Reveals New Loci for Kidney Function. PLoS Genet. 2012, 8, e1002584. [Google Scholar] [CrossRef]
- Jupp, S.; Klein, J.; Schanstra, J.; Stevens, R. Developing a kidney and urinary pathway knowledge base. J. Biomed. Semant. 2011, 2, S7. [Google Scholar] [CrossRef]
- Fernandes, M.; Husi, H. Establishment of a integrative multi-omics expression database CKDdb in the context of chronic kidney disease (CKD). Sci. Rep. 2017, 7, 40367. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Zhong, Q.; Simonis, N.; Li, Q.-R.; Charloteaux, B.; Heuze, F.; Klitgord, N.; Tam, S.; Yu, H.; Venkatesan, K.; Mou, D.; et al. Edgetic perturbation models of human inherited disorders. Mol. Syst. Biol. 2009, 5, 321. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Varkonyi, P.; Muresan, S. Complementarity Between Public and Commercial Databases: New Opportunities in Medicinal Chemistry Informatics. Curr. Top. Med. Chem. 2007, 7, 1502–1508. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef]
- He, Y.; Xiang, Z.; Zheng, J.; Lin, Y.; Overton, J.A.; Ong, E. The eXtensible ontology development (XOD) principles and tool implementation to support ontology interoperability. J. Biomed. Semant. 2018, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Snyder, M.P.; et al. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283.e19. [Google Scholar] [CrossRef]
- Digre, A.; Lindskog, C. The human protein atlas—Integrated omics for single cell mapping of the human proteome. Protein Sci. 2023, 32, e4562. [Google Scholar] [CrossRef]
- Lam, K.B.; Faust, K.; Yin, R.; Fiala, C.; Diamandis, P. The Brain Protein Atlas: A conglomerate of proteomics datasets of human neural tissue. Proteomics 2022, 22, e2200127. [Google Scholar] [CrossRef]
- Yamamoto, T. The 4th Human Kidney and Urine Proteome Project (HKUPP) Workshop 26 September 2009, Toronto, Canada. Proteomics 2010, 10, 2069–2070. [Google Scholar] [CrossRef]
- Lysenko, A.; Roznovăţ, I.A.; Saqi, M.; Mazein, A.; Rawlings, C.J.; Auffray, C. Representing and querying disease networks using graph databases. BioData Min. 2016, 9, 23. [Google Scholar] [CrossRef]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, E.; Nováček, V.; Vandenbussche, P.-Y. Facilitating prediction of adverse drug reactions by using knowledge graphs and multi-label learning models. Brief. Bioinform. 2019, 20, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Bean, D.M.; Wu, H.; Iqbal, E.; Dzahini, O.; Ibrahim, Z.M.; Broadbent, M.; Stewart, R.; Dobson, R.J.B. Knowledge graph prediction of unknown adverse drug reactions and validation in electronic health records. Sci. Rep. 2017, 7, 16416. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Masilamani, V.; Mukherjee, A. A knowledge graph embedding based approach to predict the adverse drug reactions using a deep neural network. J. Biomed. Inform. 2022, 132, 104122. [Google Scholar] [CrossRef]
- Bobed, C.; Douze, L.; Ferré, S.; Marcilly, R. PEGASE: A Knowledge Graph for Search and Exploration in Pharmacovigilance Data. In Proceedings of the EKAW Posters and Demonstrations et EKAW 2018—21st International Conference on Knowledge Engineering and Knowledge Management, Nancy, France, 12–16 November 2018; Available online: https://hal.inria.fr/hal-01976818 (accessed on 14 January 2023).
- Soldatos, T.G.; Taglang, G.; Jackson, D.B. In Silico Profiling of Clinical Phenotypes for Human Targets Using Adverse Event Data. High Throughput. 2018, 7, 37. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef]
- The ATC Ontology [WHOCC—Structure and Principles. Available online: https://www.whocc.no/atc/structure_and_principles/ (accessed on 19 December 2022).
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Van Vleet, T.R. Novel Computational Approach to Predict Off-Target Interactions for Small Molecules. Front. Big Data 2019, 2, 25. [Google Scholar] [CrossRef]
- The OFF-X Platform. Available online: https://clarivate.com/products/biopharma/off-x (accessed on 8 January 2023).
- Kim, S.; Lahu, G.; Vakilynejad, M.; Soldatos, T.G.; Jackson, D.B.; Lesko, L.J.; Trame, M.N. A case study of a patient-centered reverse translational systems-based approach to understand adverse event profiles in drug development. Clin. Transl. Sci. 2022, 15, 1003–1013. [Google Scholar] [CrossRef]
- Force, T.; Krause, D.S.; Van Etten, R.A. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 2007, 7, 332–344. [Google Scholar] [CrossRef]
- Grazette, L.P.; Boecker, W.; Matsui, T.; Semigran, M.; Force, T.; Hajjar, R.J.; Rosenzweig, A. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: Implications for herceptin-induced cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2231–2238. [Google Scholar] [CrossRef]
- Kim, S.; Lahu, G.; Vakilynejad, M.; Soldatos, T.G.; Jackson, D.B.; Lesko, L.J.; Trame, M.N. Application of a patient-centered reverse translational systems-based approach to understand mechanisms of an adverse drug reaction of immune checkpoint inhibitors. Clin. Transl. Sci. 2022, 15, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Tafenoquine Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/210795s001lbl.pdf (accessed on 18 January 2023).
- Monteluskast Label. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-boxed-warning-about-serious-mental-health-side-effects-asthma-and-allergy-drug (accessed on 19 January 2023).
- Racz, R.; Soldatos, T.G.; Jackson, D.; Burkhart, K. Association Between Serotonin Syndrome and Second-Generation Antipsychotics via Pharmacological Target-Adverse Event Analysis. Clin. Transl. Sci. 2018, 11, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Schotland, P.; Racz, R.; Jackson, D.; Levin, R.; Strauss, D.G.; Burkhart, K. Target-Adverse Event Profiles to Augment Pharmacovigilance: A Pilot Study with Six New Molecular Entities. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Schotland, P.; Racz, R.; Jackson, D.B.; Soldatos, T.G.; Levin, R.; Strauss, D.G.; Burkhart, K. Target Adverse Event Profiles for Predictive Safety in the Postmarket Setting. Clin. Pharmacol. Ther. 2021, 109, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Daluwatte, C.; Schotland, P.; Strauss, D.G.; Burkhart, K.K.; Racz, R. Predicting potential adverse events using safety data from marketed drugs. BMC Bioinform. 2020, 21, 163. [Google Scholar] [CrossRef] [PubMed]
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by β-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013, 4, 1403. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, A.N.; Castaneda, I.; Hezel, A.F.; Bascomb, N.F.; Bhattacharyya, G.S.; Abou-Alfa, G.K. Effect of coadministration of propranolol and etodolac (VT-122) plus sorafenib for patients with advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2015, 33, 390. [Google Scholar] [CrossRef]
- Srinivasan, A. Propranolol: A 50-year historical perspective. Ann. Indian Acad. Neurol. 2019, 22, 21–26. [Google Scholar] [CrossRef]
- Fjæstad, K.Y.; Rømer, A.M.A.; Goitea, V.; Johansen, A.Z.; Thorseth, M.-L.; Carretta, M.; Engelholm, L.H.; Grøntved, L.; Junker, N.; Madsen, D.H. Blockade of beta-adrenergic receptors reduces cancer growth and enhances the response to anti-CTLA4 therapy by modulating the tumor microenvironment. Oncogene 2022, 41, 1364–1375. [Google Scholar] [CrossRef]
- Amaya, C.N.; Perkins, M.; Belmont, A.; Herrera, C.; Nasrazadani, A.; Vargas, A.; Khayou, T.; Montoya, A.; Ballou, Y.; Galvan, D.; et al. Non-selective beta blockers inhibit angiosarcoma cell viability and increase progression free- and overall-survival in patients diagnosed with metastatic angiosarcoma. Oncoscience 2018, 5, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Shaghaghi, Z.; Alvandi, M.; Farzipour, S.; Dehbanpour, M.R.; Nosrati, S. A review of effects of atorvastatin in cancer therapy. Med. Oncol. 2022, 40, 27. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.; Zhang, Z.; Tan, Y.; Zhang, X.; Wei, X.; Zhu, Q. Repurposing antifungal drugs for cancer therapy. J. Adv. Res. 2023, 48, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a Commonly Used Antifungal that Inhibits Hedgehog Pathway Activity and Cancer Growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef]
- Cheng, F.; Liu, C.; Jiang, J.; Lu, W.; Li, W.; Liu, G.; Zhou, W.; Huang, J.; Tang, Y. Prediction of Drug-Target Interactions and Drug Repositioning via Network-Based Inference. PLOS Comput. Biol. 2012, 8, e1002503. [Google Scholar] [CrossRef]
- Choi, C.H.; Ryu, J.-Y.; Cho, Y.-J.; Jeon, H.-K.; Choi, J.-J.; Ylaya, K.; Lee, Y.-Y.; Kim, T.-J.; Chung, J.-Y.; Hewitt, S.M.; et al. The anti-cancer effects of itraconazole in epithelial ovarian cancer. Sci. Rep. 2017, 7, 6552. [Google Scholar] [CrossRef]
- Huang, L.; Injac, S.G.; Cui, K.; Braun, F.; Lin, Q.; Du, Y.; Zhang, H.; Kogiso, M.; Lindsay, H.; Zhao, S.; et al. Systems biology–based drug repositioning identifies digoxin as a potential therapy for groups 3 and 4 medulloblastoma. Sci. Transl. Med. 2018, 10, eaat0150. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, M.; Jia, P.; Wang, L.; Wu, Y.; Iverson, C.; Zhou, Y.; Bowton, E.; Roden, D.M.; Denny, J.C.; et al. Deciphering Signaling Pathway Networks to Understand the Molecular Mechanisms of Metformin Action. PLoS Comput. Biol. 2015, 11, e1004202. [Google Scholar] [CrossRef]
- Sheridan, C. Massive data initiatives and AI provide testbed for pandemic forecasting. Nat. Biotechnol. 2020, 38, 1010–1013. [Google Scholar] [CrossRef]
- Domingo-Fernández, D.; Baksi, S.; Schultz, B.; Gadiya, Y.; Karki, R.; Raschka, T.; Ebeling, C.; Hofmann-Apitius, M.; Kodamullil, A.T. COVID-19 Knowledge Graph: A computable, multi-modal, cause-and-effect knowledge model of COVID-19 pathophysiology. Bioinformatics 2021, 37, 1332–1334. [Google Scholar] [CrossRef]
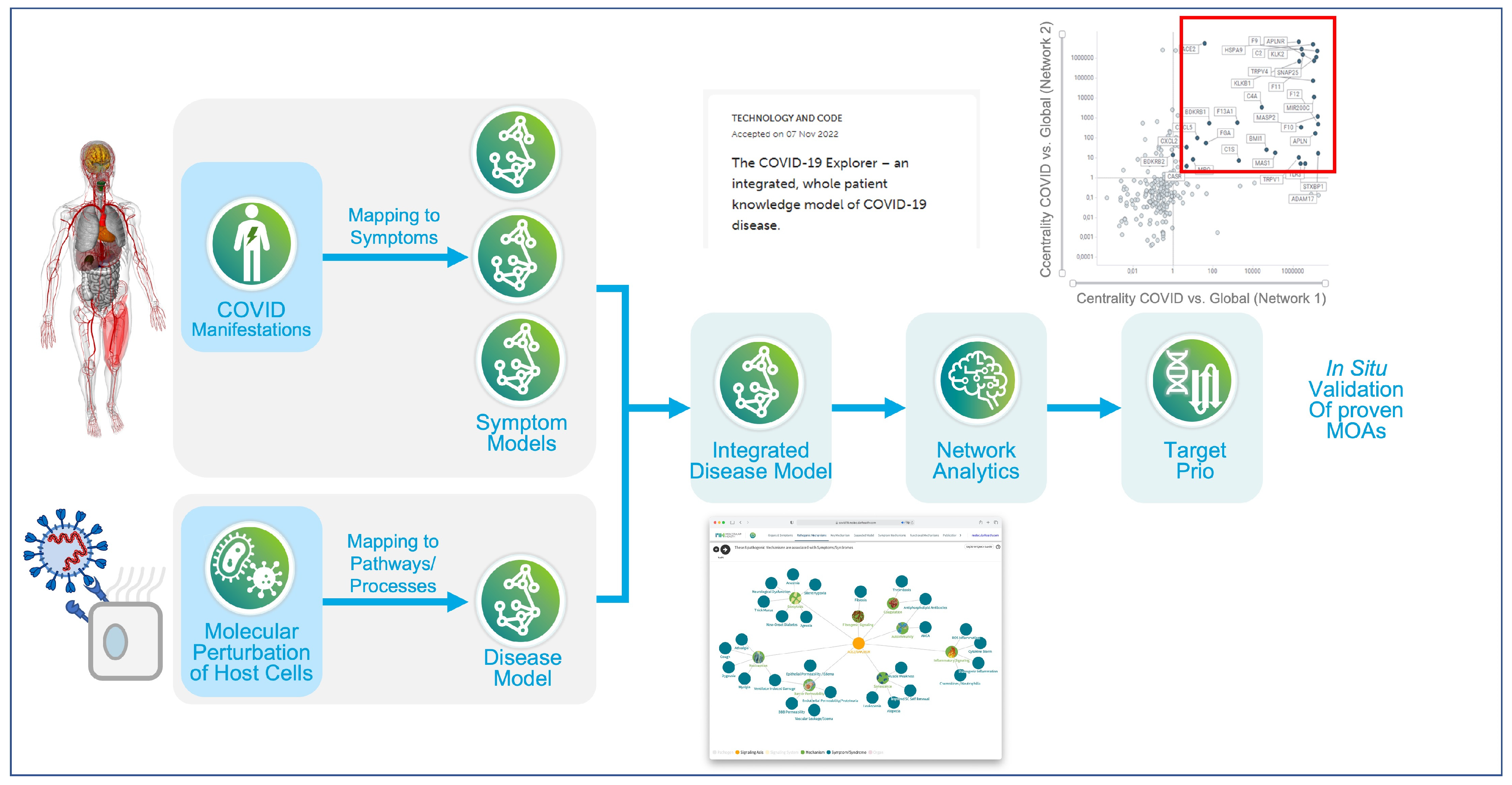
- Brock, S.; Soldatos, T.G.; Jackson, D.B.; Diella, F.; Hornischer, K.; Schäfer, A.; Hoerstrup, S.P.; Emmert, M.Y. The COVID-19 explorer—An integrated, whole patient knowledge model of COVID-19 disease. Front. Mol. Med. 2022, 2, 1035215. [Google Scholar] [CrossRef]
- Brock, S.; Jackson, D.B.; Soldatos, T.G.; Hornischer, K.; Schäfer, A.; Diella, F.; Emmert, M.Y.; Hoerstrup, S.P. Whole patient knowledge modeling of COVID-19 symptomatology reveals common molecular mechanisms. Front. Mol. Med. 2022, 2, 1035290. [Google Scholar] [CrossRef]
- Ringel, M.S.; Scannell, J.W.; Baedeker, M.; Schulze, U. Breaking Eroom’s Law. Nat. Rev. Drug Discov. 2020, 19, 833–834. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Bosley, J. When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis. PLoS ONE 2016, 11, e0147215. [Google Scholar] [CrossRef]
- Morgan, P.; Brown, D.G.; Lennard, S.; Anderton, M.J.; Barrett, J.C.; Eriksson, U.; Fidock, M.; Hamrén, B.; Johnson, A.; March, R.E.; et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat. Rev. Drug Discov. 2018, 17, 167–181. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- 230 AI-Driven Drug Discovery Start-Ups. Available online: https://blog.benchsci.com/startups-using-artificial-intelligence-in-drug-discovery (accessed on 2 December 2022).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, D.B.; Racz, R.; Kim, S.; Brock, S.; Burkhart, K. Rewiring Drug Research and Development through Human Data-Driven Discovery (HD3). Pharmaceutics 2023, 15, 1673. https://doi.org/10.3390/pharmaceutics15061673
Jackson DB, Racz R, Kim S, Brock S, Burkhart K. Rewiring Drug Research and Development through Human Data-Driven Discovery (HD3). Pharmaceutics. 2023; 15(6):1673. https://doi.org/10.3390/pharmaceutics15061673
Chicago/Turabian StyleJackson, David B., Rebecca Racz, Sarah Kim, Stephan Brock, and Keith Burkhart. 2023. "Rewiring Drug Research and Development through Human Data-Driven Discovery (HD3)" Pharmaceutics 15, no. 6: 1673. https://doi.org/10.3390/pharmaceutics15061673
APA StyleJackson, D. B., Racz, R., Kim, S., Brock, S., & Burkhart, K. (2023). Rewiring Drug Research and Development through Human Data-Driven Discovery (HD3). Pharmaceutics, 15(6), 1673. https://doi.org/10.3390/pharmaceutics15061673