
Prediction of CYP-Mediated Drug Interaction Using Physiologically Based Pharmacokinetic Modeling: A Case Study of Salbutamol and Fluvoxamine



Abstract
1. Introduction
2. Materials and Methods
2.1. Prediction of Pharmacokinetic and Physicochemical Properties of Salbutamol
2.2. Screening of Potential Drug Interactors with Salbutamol
2.3. PBPK Modeling Development
2.4. PBPK Model Validation
2.5. DDI Simulation between Salbutamol and Fluvoxamine
3. Results and Discussion
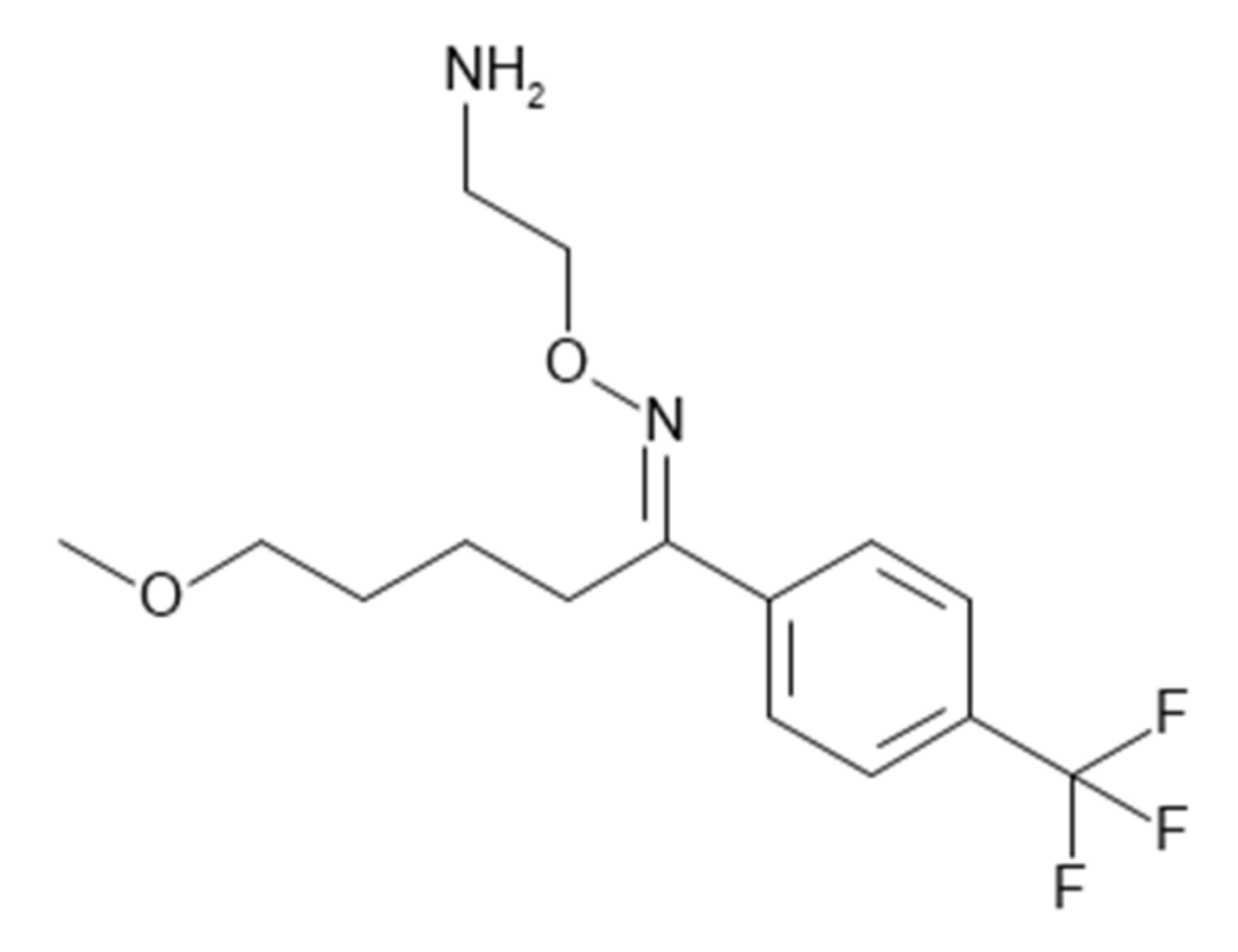
3.1. Fluvoxamine as the Perpetrator Drug for Salbutamol DDI Study
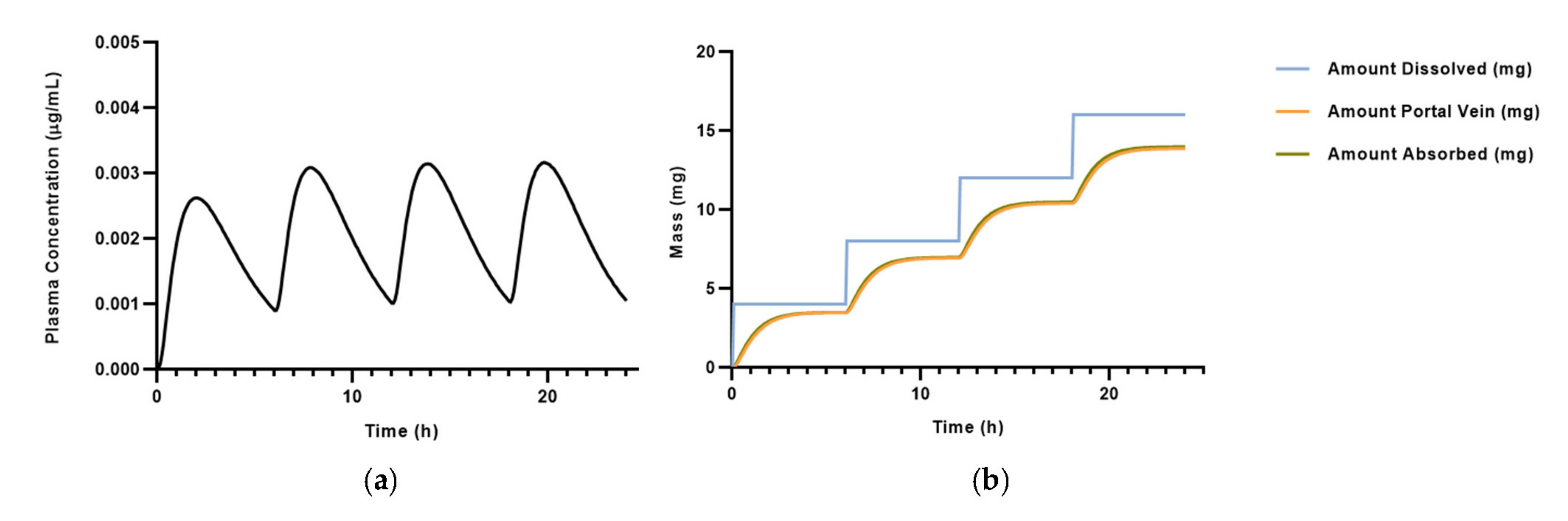
3.2. PBPK Model for Salbutamol
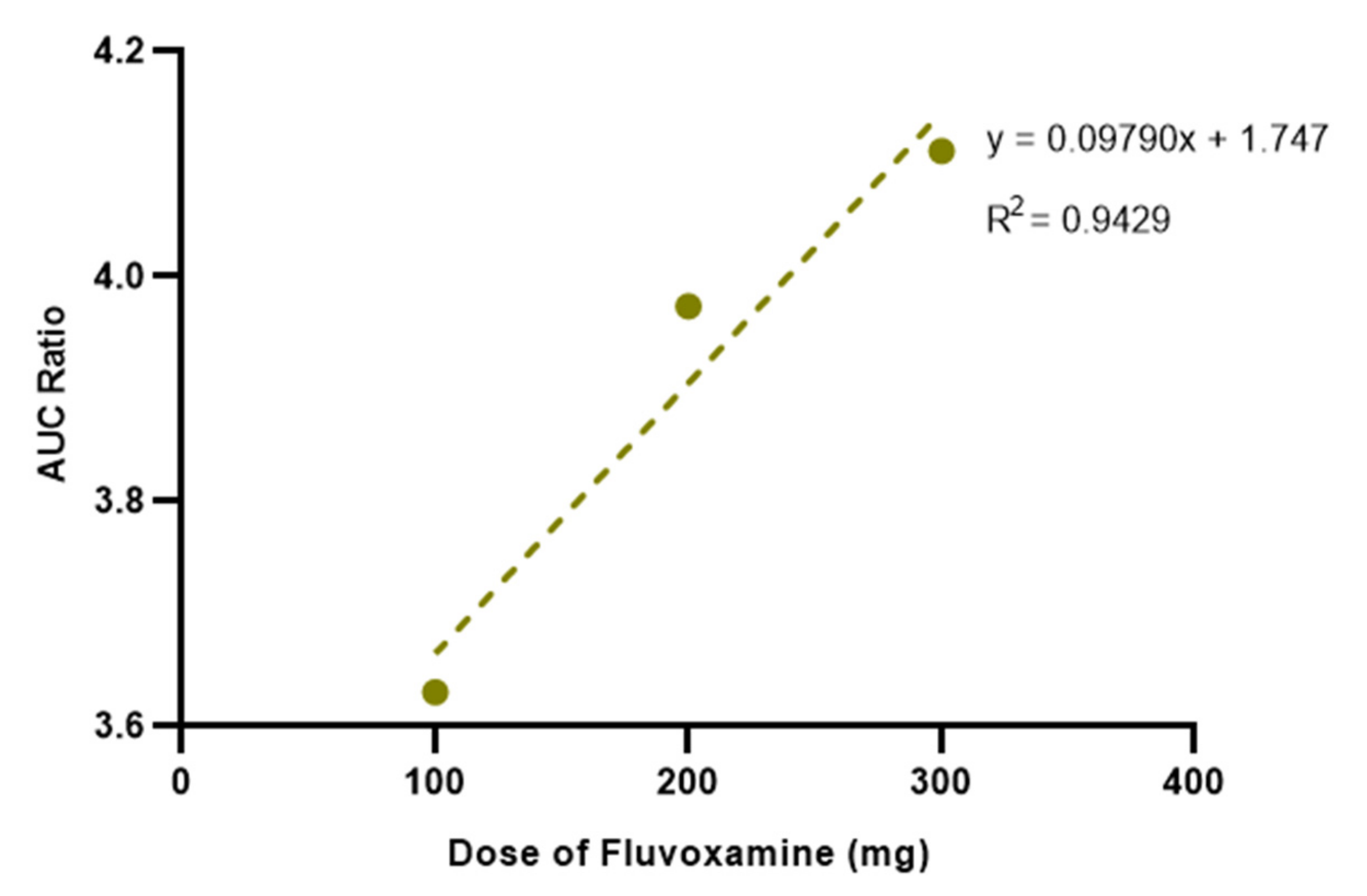
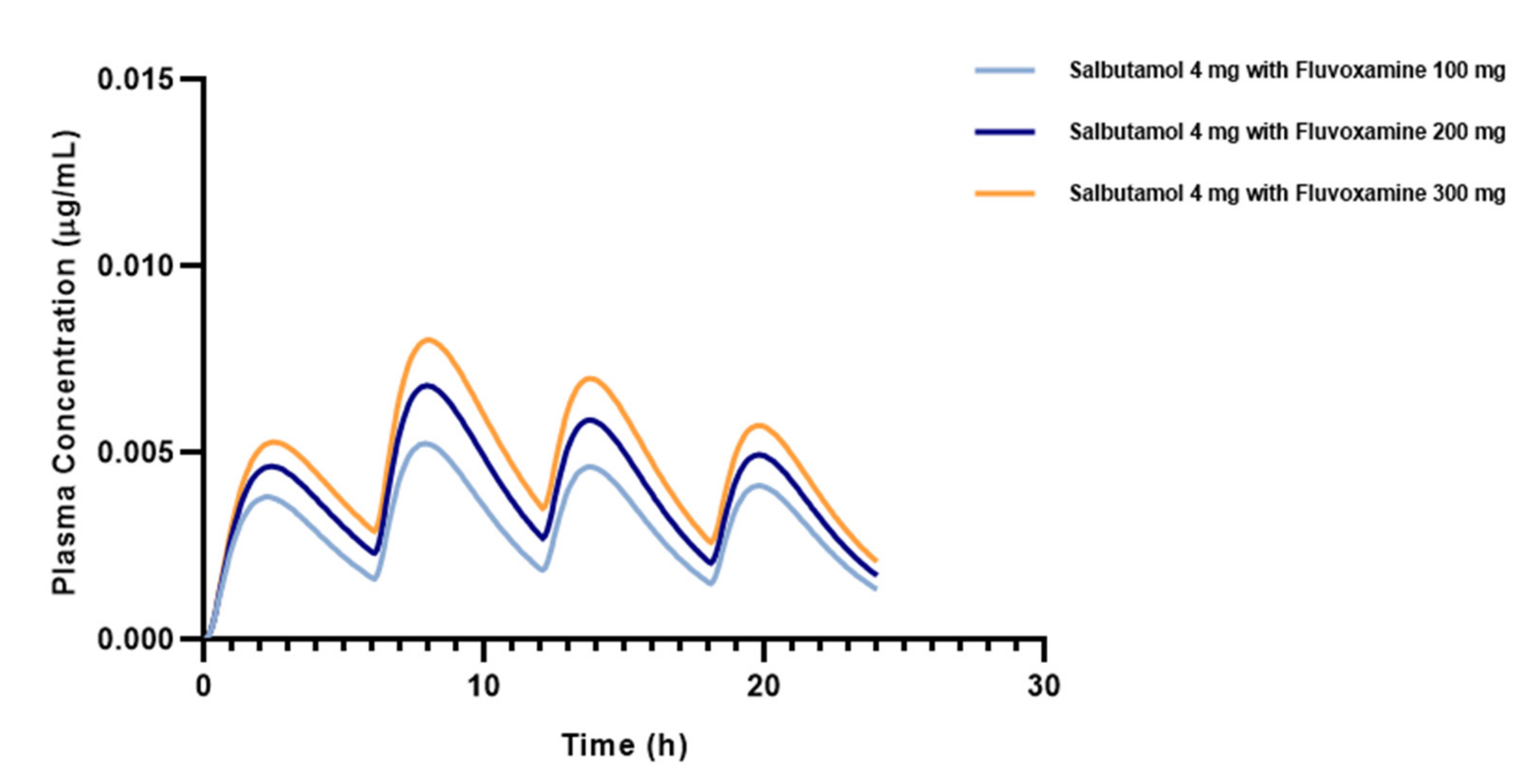
3.3. Effect of Different Doses of Fluvoxamine on Salbutamol Pharmacokinetics
3.4. Effect of Different Ages on Salbutamol Pharmacokinetics Co-Administered with Fluvoxamine
3.5. Effect of Comorbidities on Salbutamol Pharmacokinetics Co-Administered with Fluvoxamine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Burke, J.P.; Classen, D.C.; Pestotnik, S.L.; Evans, R.S.; Lloyd, J.F. Adverse Drug Events in Hospitalized Patients-Reply. JAMA J. Am. Med. Assoc. 1997, 277, 1353. [Google Scholar] [CrossRef]
- Han, K.; Cao, P.; Wang, Y.; Xie, F.; Ma, J.; Yu, M.; Wang, J.; Xu, Y.; Zhang, Y.; Wan, J. A Review of Approaches for Predicting Drug–Drug Interactions Based on Machine Learning. Front. Pharmacol. 2022, 12, 814858. [Google Scholar] [CrossRef] [PubMed]
- Hohl, C.M.; Dankoff, J.; Colacone, A.; Afilalo, M. Polypharmacy, Adverse Drug-Related Events, and Potential Adverse Drug Interactions in Elderly Patients Presenting to an Emergency Department. Ann. Emerg. Med. 2001, 38, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J. Drug Interaction and Pharmacist. J. Young Pharm. 2010, 2, 326–331. [Google Scholar] [CrossRef]
- Roberts, A.G.; Gibbs, M.E. Mechanisms and the Clinical Relevance of Complex Drug–Drug Interactions. Clin. Pharmacol. Adv. Appl. 2018, 10, 123–134. [Google Scholar] [CrossRef]
- Peng, Y.; Cheng, Z.; Xie, F. Evaluation of Pharmacokinetic Drug-Drug Interactions: A Review of the Mechanisms, in Vitro and in Silico Approaches. Metabolites 2021, 11, 75. [Google Scholar] [CrossRef]
- Niu, J.; Straubinger, R.M.; Mager, D.E. Pharmacodynamic Drug–Drug Interactions. Clin. Pharmacol. Ther. 2019, 105, 1395–1406. [Google Scholar] [CrossRef]
- Manzi, S.F.; Shannon, M. Drug Interactions—A Review. Clin. Pediatr. Emerg. Med. 2005, 6, 93–102. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2017, 19, 38–54. [Google Scholar] [CrossRef]
- Rowland, M.; Tozer, T. Clinical Pharmacokinetics and Pharmacodynamics; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- Spina, E.; Santoro, V.; D’Arrigo, C. Clinically Relevant Pharmacokinetic Drug Interactions with Second-Generation Antidepressants: An Update. Clin. Ther. 2008, 30, 1206–1227. [Google Scholar] [CrossRef]
- Reddel, H.K.; Bacharier, L.B.; Bateman, E.D.; Brightling, C.E.; Brusselle, G.G.; Buhl, R.; Cruz, A.A.; Duijts, L.; Drazen, J.M.; FitzGerald, J.M.; et al. Global Initiative for Asthma Strategy 2021: Executive Summary and Rationale for Key Changes. Eur. Respir. J. 2022, 59, 2102730. [Google Scholar] [CrossRef] [PubMed]
- 2022 GINA Main Report—Global Initiative for Asthma—GINA. Available online: https://ginasthma.org/gina-reports/ (accessed on 9 March 2023).
- Ledford, D.K.; Lockey, R.F. Asthma and Comorbidities. Curr. Opin. Allergy Clin. Immunol. 2013, 13, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Liu, S.; Yang, Y.; Zhao, T.; Li, S.; Zhou, T.; Tan, W. Identification of the Major Metabolites of (R)-Salbutamol in Human Urine, Plasma and Feces Using Ultra High Performance Liquid Chromatography Coupled with Quadrupole Time-of-Flight Mass Spectrometry. J. Sep. Sci. 2019, 42, 3200–3208. [Google Scholar] [CrossRef] [PubMed]
- Schmekel, B.; Rydberg, I.; Norlander, B.; Sjöswärd, K.N.; Ahlner, J.; Andersson, R.G.G. Stereoselective Pharmacokinetics of S-Salbutamol after Administration of the Racemate in Healthy Volunteers. Eur. Respir. J. 1999, 13, 1230–1235. [Google Scholar] [CrossRef]
- Ahrens, R.C.; Smith, G.D.; Pharm, D. Albuterol: An Adrenergic Agent for Use in the Treatment of Asthma Pharmacology, Pharmacokinetics and Clinical Use. Pharmacol. Pharm. 1984, 4, 105–120. [Google Scholar] [CrossRef]
- Salbutamol: Uses, Interactions, Mechanism of Action DrugBank Online. Available online: https://go.drugbank.com/drugs/DB01001 (accessed on 14 January 2023).
- Marques, L.; Vale, N. Unraveling the Impact of Salbutamol Polytherapy: Clinically Relevant Drug Interactions. Futur. Pharmacol. 2023, 3, 19. [Google Scholar] [CrossRef]
- Albuterol—Drug Usage Statistics, ClinCalc DrugStats Database. Available online: https://clincalc.com/DrugStats/Drugs/Albuterol (accessed on 14 May 2023).
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Salbutamol: Uses, Dosage, Side Effects, Warnings—Drugs.Com. Available online: https://www.drugs.com/salbutamol.html (accessed on 10 October 2022).
- Salbutamol C13H21NO3—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Salbutamol (accessed on 22 April 2023).
- COVID-19 Treatment Guidelines Panel Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 22 April 2023).
- Reis, G.; dos Santos Moreira-Silva, E.A.; Silva, D.C.M.; Thabane, L.; Milagres, A.C.; Ferreira, T.S.; dos Santos, C.V.Q.; de Souza Campos, V.H.; Nogueira, A.M.R.; de Almeida, A.P.F.G.; et al. Effect of Early Treatment with Fluvoxamine on Risk of Emergency Care and Hospitalisation among Patients with COVID-19: The TOGETHER Randomised, Platform Clinical Trial. Lancet Glob. Heal. 2022, 10, e42–e51. [Google Scholar] [CrossRef]
- Marzolini, C.; Marra, F.; Boyle, A.; Khoo, S.; Back, D.J. Fluvoxamine for the Treatment of COVID-19. Lancet Glob. Health 2022, 10, e331. [Google Scholar] [CrossRef]
- Nyirenda, J.L.Z.; Sofroniou, M.; Toews, I.; Mikolajewska, A.; Lehane, C.; Monsef, I.; Abu-taha, A.; Maun, A.; Stegemann, M.; Schmucker, C. Fluvoxamine for the Treatment of COVID-19. Cochrane Database Syst. Rev. 2022, 9, CD015391. [Google Scholar] [CrossRef] [PubMed]
- Crewe, H.K.; Lennard, M.S.; Tucker, G.T.; Woods, F.R.; Haddock, R.E.; Brøsen, K. The Effect of Selective Serotonin Re-Uptake Inhibitors on Cytochrome P4502D6 (CYP2D6) Activity in Human Liver Microsomes. Br. J. Clin. Pharmacol. 2004, 58, S744–S747. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Menke, J.J. Drug Interactions and the Cytochrome P-450 System. S. D. J. Med. 2000, 53, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Van Harten, J. Overview of the Pharmacokinetics of Fluvoxamine. Clin. Pharmacokinet. 1995, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brosen, K.; Skjelbo, E.; Rasmussen, B.B.; Poulsen, H.E.; Loft, S. Fluvoxamine Is a Potent Inhibitor of Cytochrome P4501A2. Biochem. Pharmacol. 1993, 45, 1211–1214. [Google Scholar] [CrossRef]
- Morgan, D.; Paull, J.; Richmond, B.; Wilson-Evered, E.; Ziccone, S. Pharmacokinetics of Intravenous and Oral Salbutamol and Its Sulphate Conjugate. Br. J. Clin. Pharmacol. 1986, 22, 587–593. [Google Scholar] [CrossRef]
- Fluvoxamine: Uses, Interactions, Mechanism of Action DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00176 (accessed on 22 April 2023).
- Drenth-van Maanen, A.C.; Wilting, I.; Jansen, P.A.F. Prescribing Medicines to Older People—How to Consider the Impact of Ageing on Human Organ and Body Functions. Br. J. Clin. Pharmacol. 2020, 86, 1921–1930. [Google Scholar] [CrossRef]
- Morselli, P.L.; Franco-Morselli, R.; Bossi, L. Clinical Pharmacokinetics in Newborns and Infants: Age-Related Differences and Therapeutic Implications. Clin. Pharmacokinet. 1980, 5, 485–527. [Google Scholar] [CrossRef]
- O’Hara, K. Pharmacokinetic Changes with Growth and Development between Birth and Adulthood. J. Pharm. Pract. Res. 2017, 47, 313–318. [Google Scholar] [CrossRef]
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental Pharmacology—Drug Disposition, Action, and Therapy in Infants and Children. N. Engl. J. Med. 2003, 12, 349. [Google Scholar] [CrossRef]
- Anderson, G.D. Children versus Adults: Pharmacokinetic and Adverse-Effect Differences. Epilepsia 2002, 43, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Waring, R.H.; Harris, R.M.; Mitchell, S.C. Drug Metabolism in the Elderly: A Multifactorial Problem? Maturitas 2017, 100, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Kinirons, M.T.; O’Mahony, M.S. Drug Metabolism and Ageing. Br. J. Clin. Pharmacol. 2004, 57, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, D.L.; Woodhouse, K.W.; Wang, R.K.; Wynne, H.; James, O.F.; McManus, M.; Kremers, P. Effects of Age and Gender on in Vitro Properties of Human Liver Microsomal Monooxygenases. Clin. Pharmacol. Ther. 1990, 48, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Sotaniemi, E.A.; Arranto, A.J.; Pelkonen, O.; Pasanen, M. Age and Cytochrome P450-Linked Drug Metabolism in Humans: An Analysis of 226 Subjects with Equal Histopathologic Conditions. Clin. Pharmacol. Ther. 1997, 61, 331–339. [Google Scholar] [CrossRef]
- Tomankova, V.; Anzenbacher, P.; Anzenbacherova, E. Effects of Obesity on Liver Cytochromes P450 in Various Animal Models. Biomed. Pap. 2017, 161, 144–151. [Google Scholar] [CrossRef]
- Brill, M.J.E.; Diepstraten, J.; Van Rongen, A.; Van Kralingen, S.; Van Den Anker, J.N.; Knibbe, C.A.J. Impact of Obesity on Drug Metabolism and Elimination in Adults and Children. Clin. Pharmacokinet. 2012, 51, 277–304. [Google Scholar] [CrossRef]
- Déri, M.T.; Kiss, Á.F.; Tóth, K.; Paulik, J.; Sárváry, E.; Kóbori, L.; Monostory, K. End-Stage Renal Disease Reduces the Expression of Drug-Metabolizing Cytochrome P450s. Pharmacol. Rep. 2020, 72, 1695–1705. [Google Scholar] [CrossRef]
- The First Trimester Johns Hopkins Medicine. Available online: https://www.hopkinsmedicine.org/health/wellness-and-prevention/the-first-trimester (accessed on 14 May 2023).
- Jeong, H. Altered Drug Metabolism during Pregnancy: Hormonal Regulation of Drug-Metabolizing Enzymes. Expert Opin. Drug Metab. Toxicol. 2010, 6, 689–699. [Google Scholar] [CrossRef]
- Fluvoxamine Use During Pregnancy Drugs.Com. Available online: https://www.drugs.com/pregnancy/fluvoxamine.html (accessed on 23 April 2023).
- Salbutamol: Inhaler to Relieve Asthma and Breathlessness—NHS. Available online: https://www.nhs.uk/medicines/salbutamol-inhaler/ (accessed on 23 April 2023).
- Anderson, G.D. Pregnancy-Induced Changes in Pharmacokinetics: A Mechanistic-Based Approach. Clin. Pharmacokinet. 2005, 44, 989–1008. [Google Scholar] [CrossRef]
- Gandhi, M.; Aweeka, F.; Greenblatt, R.M.; Blaschke, T.F. Sex Differences in Pharmacokinetics and Pharmacodynamics. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 499–523. [Google Scholar] [CrossRef] [PubMed]
- Waxman, D.J.; Holloway, M.G. Sex Differences in the Expression of Hepatic Drug Metabolizing Enzymes. Mol. Pharmacol. 2009, 76, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Gender Differences in Pharmacokinetics. Available online: https://www.uspharmacist.com/article/gender-differences-in-pharmacokinetics (accessed on 23 April 2023).
- Rubinow, D.R.; Moore, M. Sex-Dependent Modulation of Treatment Response. Dialogues Clin. Neurosci. 2004, 6, 39–51. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
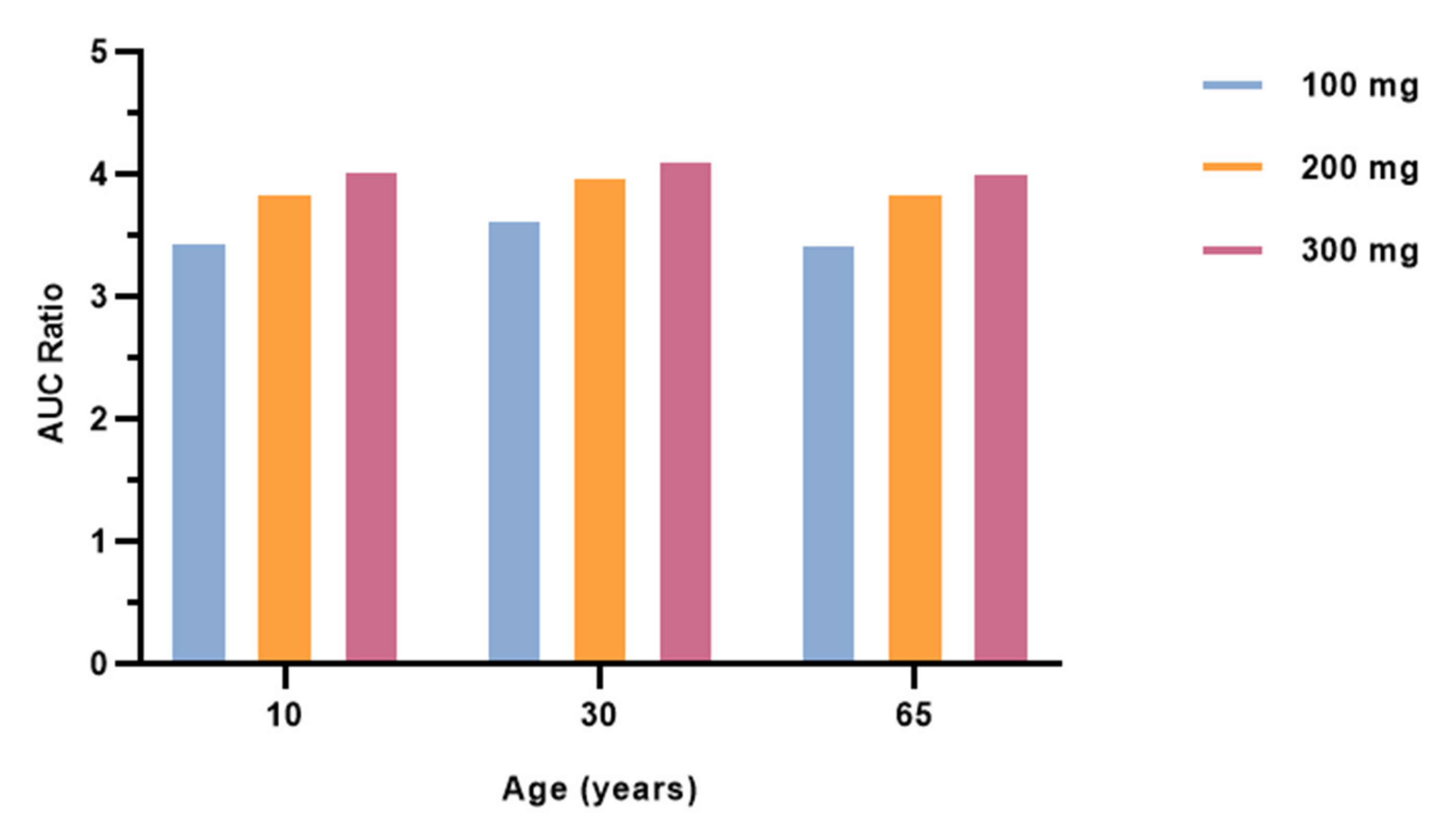{kind=link}
| Physicochemical Properties | Predicted Value | Optimized Value | Reference |
|---|---|---|---|
| Log P | 1.644 | 1.4 | [18,21,22,23,24] |
| Ionization constant (pKa) | 9.98 | 10.3 | |
| Molecular Weight (g/mol) | 239.317 | 239.31 | |
| Water Solubility (mg/mL) | 15.869 | 9.53 | [22] |
| Diff. Coeff. (cm2/s·105) | 0.804 | ND | ND |
| Peff (cm/s·104) | 1.331 | 0.815 | Calculated from pkCSM [21] |
| BBB penetration | Low | Low | Calculated from SwissADME and pkCSM [21,22] |
| Drug | CYP Enzyme | Inhibitor | Substrate | Km | Vmax | CL | Sites of Metabolism |
|---|---|---|---|---|---|---|---|
| Salbutamol | 1A2 | No (90%) | No (97%) | NS | NS | NS | NS |
| 2A6 | ND | No (98%) | NS | NS | NS | NS | |
| 2B6 | ND | No (65%) | NS | NS | NS | NS | |
| 2C8 | ND | No (92%) | NS | NS | NS | NS | |
| 2C9 | No (99%) | No (98%) | NS | NS | NS | NS | |
| 2C19 | ND | Yes (82%) | 30.146 | 157.577 | 73.179 | C7 | |
| 2D6 | Yes (49%) | Yes (66%) | 37.808 | 2.201 | 0.466 | C17 | |
| 2E1 | ND | No (91%) | NS | NS | NS | NS | |
| 3A4 | No (78%) | No (84%) | NS | NS | NS | NS | |
| Fluvoxamine | 1A2 | No (51%) | Yes (48%) | 1.821 | 1.500 | 42.835 | C1, C11 |
| 2A6 | ND | No (82%) | NS | NS | NS | NS | |
| 2B6 | ND | No (83%) | NS | NS | NS | NS | |
| 2C8 | ND | No (99%) | NS | NS | NS | NS | |
| 2C9 | Yes (41%) | No (78%) | NS | NS | NS | NS | |
| 2C19 | No (95%) | Yes (67%) | 22.656 | 250.097 | 154.542 | C1, C3, C11, C12 | |
| 2D6 | Yes (70%) | Yes (66%) | 0.674 | 3.937 | 46.721 | C1, C3, C11 | |
| 2E1 | ND | Yes (78%) | ND | ND | ND | C1, C3, C12 | |
| 3A4 | Yes (80%) | No (54%) | NS | NS | NS | NS |
| Pharmacokinetic Parameters | Observed Value | Estimated Value |
|---|---|---|
| Fa (%) | 88.82 | 88.079 |
| FDp (%) | ND | 87.486 |
| F (%) | ND | 29.447 |
| Cmax (μg/mL) | 0.01013 | 3.159 × 10−3 |
| Tmax (h) | 2.73 | 19.84 |
| AUC0–inf (μg*h/mL) | 0.1094 | 0.05235 |
| AUC0–t (μg*h/mL) | 0.1094 | 0.04929 |
| Cmax liver (μg/mL) | ND | 7.57 × 10−3 |
| Compound | Fa (%) | FDp (%) | F (%) | Cmax (μg/mL) | Tmax (h) | AUC0–t (ng.h/mL) | AUC0-inf (ng.h/mL) |
|---|---|---|---|---|---|---|---|
| Salbutamol baseline | 88.08 | 87.49 | 29.44 | 0.0032 | 19.76 | 52.34 | 49.65 |
| Salbutamol 4 mg + fluvoxamine 100 mg | 88.06 | 87.47 | 38.50 | 0.0052 | 7.92 | 78.04 | 74.19 |
| Salbutamol 4 mg + fluvoxamine 200 mg | 88.04 | 87.45 | 44.76 | 0.0067 | 7.92 | 100.6 | 95.39 |
| Salbutamol 4 mg + fluvoxamine 300 mg | 88.03 | 87.43 | 49.41 | 0.0080 | 8.00 | 120.6 | 113.9 |
| Dosing Regimen | Concentration Type | AUC Ratio | DDI Classification | ||||
|---|---|---|---|---|---|---|---|
| Age | 10 | 30 | 65 | 10 | 30 | 65 | |
| Salbutamol with fluvoxamine 100 mg | Cmax | 3.453 | 3.630 | 3.431 | M | M | M |
| Liver Unbound | 2.726 | 2.021 | 2.586 | M | M | M | |
| Salbutamol with fluvoxamine 200 mg | Cmax | 3.854 | 3.973 | 3.839 | M | M | M |
| Liver Unbound | 3.287 | 2.558 | 3.156 | M | M | M | |
| Salbutamol with fluvoxamine 300 mg | Cmax | 4.022 | 4.111 | 4.011 | M | M | M |
| Liver Unbound | 3.567 | 2.903 | 3.456 | M | M | M | |
| Dosing Regimen | Concentration Type | AUC Ratio | DDI Classification | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Physiological Status | Healthy | OW | Obese | MildRI | MRI | SRI | Healthy | OW | Obese | MildRI | MRI | SRI | |
| Salbutamol 4 mg + fluvoxamine 100 mg | Cmax | 3.630 | 3.420 | 3.413 | 3.423 | 3.423 | 3.423 | M | M | M | M | M | M |
| Liver Unbound | 2.021 | 2.501 | 2.451 | 2.511 | 2.511 | 2.511 | M | M | M | M | M | M | |
| Salbutamol 4 mg + fluvoxamine 200 mg | Cmax | 3.973 | 3.831 | 3.827 | 3.832 | 3.832 | 3.832 | M | M | M | M | M | M |
| Liver Unbound | 2.558 | 3.075 | 3.031 | 3.089 | 3.089 | 3.089 | M | M | M | M | M | M | |
| Salbutamol 4 mg + fluvoxamine 300 mg | Cmax | 4.111 | 4.005 | 4.001 | 4.006 | 4.006 | 4.006 | M | M | M | M | M | M |
| Liver Unbound | 2.903 | 3.385 | 3.341 | 3.396 | 3.396 | 3.396 | M | M | M | M | M | M | |
| Dosing Regimen | Concentration Type | AUC Ratio | DDI Classification | ||
|---|---|---|---|---|---|
| Age | Female | Pregnant | Female | Pregnant | |
| Salbutamol 4 mg + fluvoxamine 100 mg | Cmax | 3.426 | 3.426 | M | M |
| Liver Unbound | 2.530 | 2.540 | M | M | |
| Salbutamol 4 mg + fluvoxamine 200 mg | Cmax | 3.835 | 3.835 | M | M |
| Liver Unbound | 3.112 | 3.117 | M | M | |
| Salbutamol 4 mg + fluvoxamine 300 mg | Cmax | 4.007 | 4.007 | M | M |
| Liver Unbound | 3.415 | 3.420 | M | M | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, L.; Vale, N. Prediction of CYP-Mediated Drug Interaction Using Physiologically Based Pharmacokinetic Modeling: A Case Study of Salbutamol and Fluvoxamine. Pharmaceutics 2023, 15, 1586. https://doi.org/10.3390/pharmaceutics15061586
Marques L, Vale N. Prediction of CYP-Mediated Drug Interaction Using Physiologically Based Pharmacokinetic Modeling: A Case Study of Salbutamol and Fluvoxamine. Pharmaceutics. 2023; 15(6):1586. https://doi.org/10.3390/pharmaceutics15061586
Chicago/Turabian StyleMarques, Lara, and Nuno Vale. 2023. "Prediction of CYP-Mediated Drug Interaction Using Physiologically Based Pharmacokinetic Modeling: A Case Study of Salbutamol and Fluvoxamine" Pharmaceutics 15, no. 6: 1586. https://doi.org/10.3390/pharmaceutics15061586
APA StyleMarques, L., & Vale, N. (2023). Prediction of CYP-Mediated Drug Interaction Using Physiologically Based Pharmacokinetic Modeling: A Case Study of Salbutamol and Fluvoxamine. Pharmaceutics, 15(6), 1586. https://doi.org/10.3390/pharmaceutics15061586