

Homology-Directed-Repair-Based Genome Editing in HSPCs for the Treatment of Inborn Errors of Immunity and Blood Disorders



Abstract
1. Introduction
2. Gene-Editing Nucleases
3. Mechanism of HDR

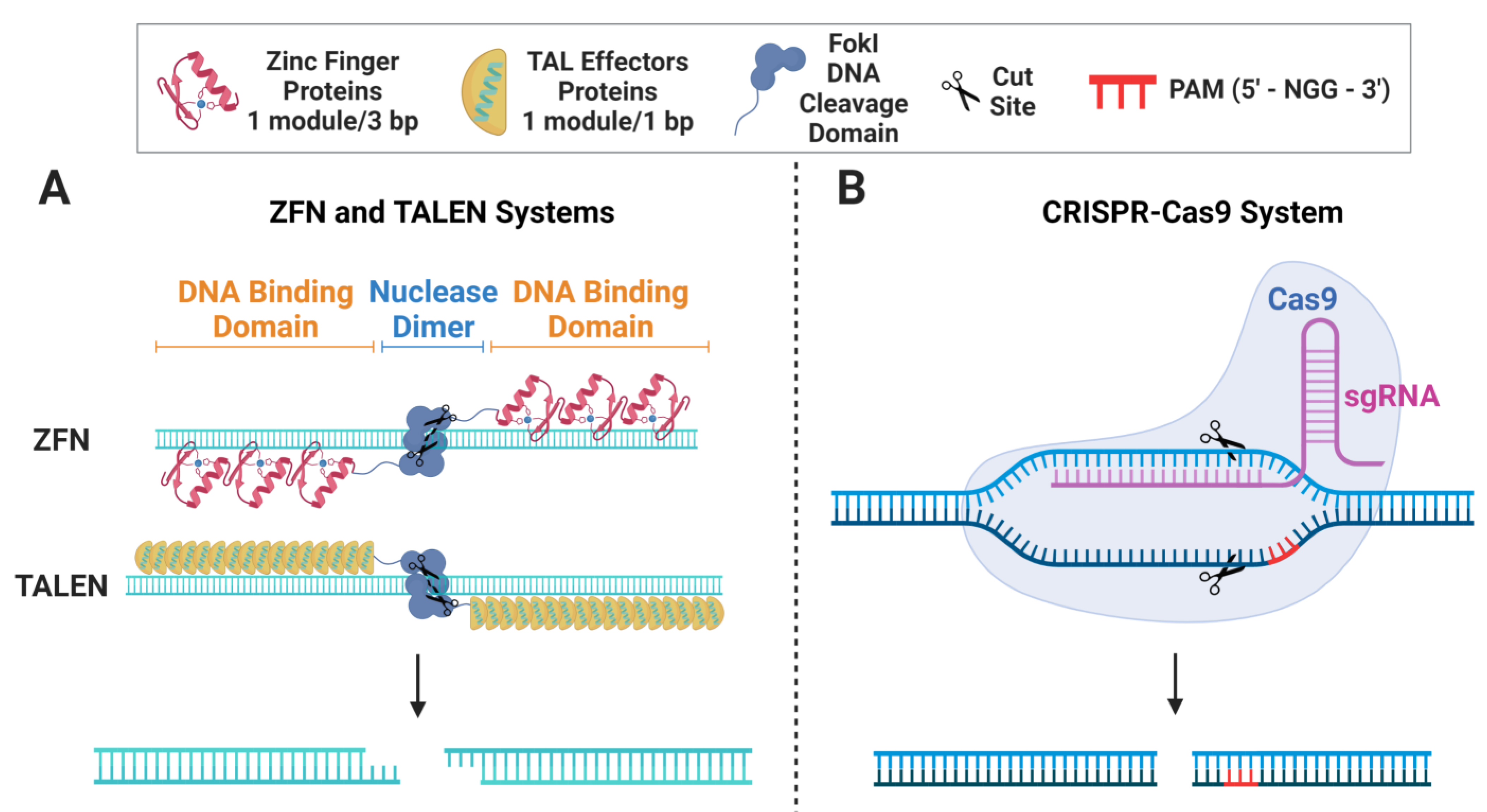{kind=link}
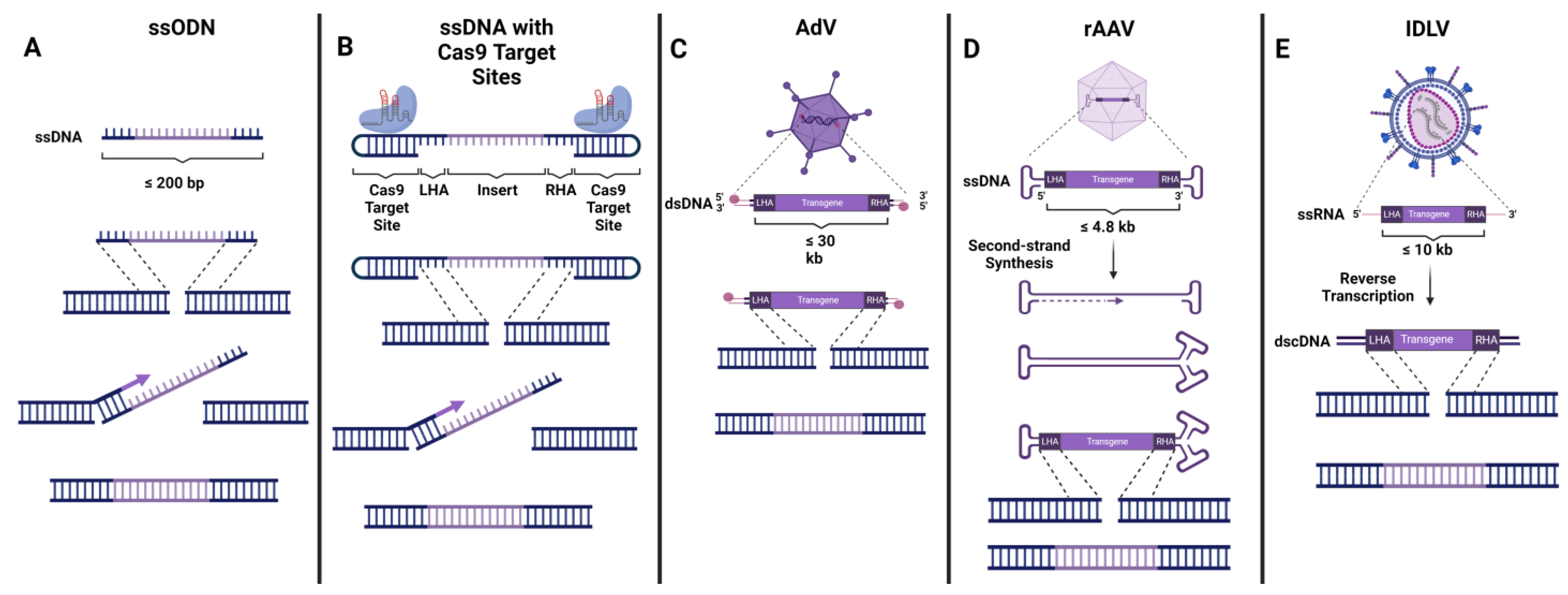{kind=link}
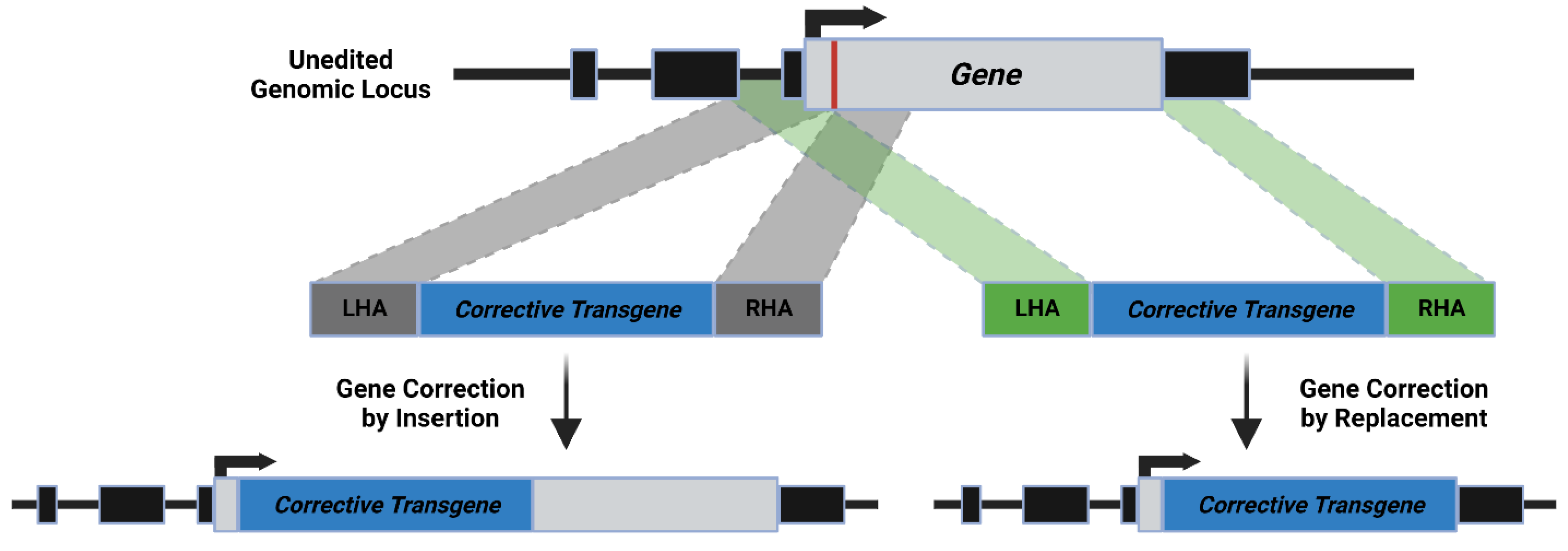{kind=link}
| Delivery Platform | Benefits | Drawbacks |
|---|---|---|
| Non-viral delivery of naked DNA |
|
|
|
| |
| ||
| AdV |
|
|
| ||
| AAV |
|
|
| ||
| ||
| IDLV |
|
|
| ||
| ||
|
4. Delivery of DNA Donor Template
4.1. Non-Viral Donor DNA Delivery
4.2. AdV
4.3. AAV
4.4. IDLV
5. Existing Barriers to Efficient HDR-Based Therapies
5.1. Specificity
5.2. Insufficient HDR/Bias towards NHEJ
5.3. Toxicity
5.4. Poor Engraftment of Edited Cells
6. Conclusions
| Targeted Gene | Nuclease | Donor Platform | Study | Reference | |
|---|---|---|---|---|---|
| SCID | WAS | Cas9 | AAV | in vivo | [32] |
| RAG2 | Cas9 | AAV | in vitro | [87,96,97] | |
| Cas9 | AAV | in vivo | [29] | ||
| IL2RG | ZFN | IDLV | in vitro | [17] | |
| ZFN | IDLV | in vivo | [103,134] | ||
| ZFN | AAV | in vivo | [134,141] | ||
| Cas9 | IDLV | in vivo | [134] | ||
| Cas9 | AAV | in vitro | [89] | ||
| Cas9 | AAV | in vivo | [30,89,134,141] | ||
| CGD | AAVS1 | Cas9 | AAV | in vivo | [90] |
| CYBB | Cas9 | ssODN | in vivo | [34] | |
| Cas9 | AAV | in vivo | [91] | ||
| NCF1 | ZFN | AAV | in vivo | [92] | |
| IPEX | FOXP3 | Cas9 | AAV | in vivo | [31] |
| SCD/ β-thalassemia | HBB | ZFN | IDLV | in vivo | [19,26] |
| ZFN | ssODN | in vitro | [23] | ||
| ZFN | ssODN | in vivo | [19,26] | ||
| ZFN | AdV | in vivo | [26] | ||
| ZFN | AAV | in vivo | [26] | ||
| TALEN | ssODN | in vitro | [23] | ||
| Cas9 | IDLV | in vitro | [24] | ||
| Cas9 | IDLV | in vivo | [26] | ||
| Cas9 | ssODN | in vitro | [23] | ||
| Cas9 | ssODN | in vivo | [22,25,26,28] | ||
| Cas9 | AAV | in vivo | [20,21,25,26,27,88] | ||
| Cas9 | AdV | in vivo | [26,79] | ||
| Hemophilia | HBA | Cas9 | AAV | in vivo | [93] |
| XHIM | CD40L | TALEN | IDLV | in vivo | [33] |
| TALEN | AAV | in vivo | [33] | ||
| Cas9 | IDLV | in vivo | [33] | ||
| Cas9 | AAV | in vivo | [33] | ||
| XLA | BTK | Cas9 | AAV | in vivo | [94] |
| XMEN | MAGT1 | Cas9 | AAV | in vivo | [95] |
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soni, S. Gene therapies for transfusion dependent β-thalassemia: Current status and critical criteria for success. Am. J. Hematol. 2020, 95, 1099–1112. [Google Scholar] [CrossRef]
- Song, R.; Zhai, Q.; Sun, L.; Huang, E.; Zhang, Y.; Zhu, Y.; Guo, Q.; Tian, Y.; Zhao, B.; Lu, H. CRISPR/Cas9 genome editing technology in filamentous fungi: Progress and perspective. Appl. Microbiol. Biotechnol. 2019, 103, 6919–6932. [Google Scholar] [CrossRef]
- Munoz, D.M.; Cassiani, P.J.; Li, L.; Billy, E.; Korn, J.M.; Jones, M.D.; Golji, J.; Ruddy, D.A.; Yu, K.; McAllister, G.; et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 2016, 6, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef]
- Sid, H.; Schusser, B. Applications of Gene Editing in Chickens: A New Era Is on the Horizon. Front. Genet. 2018, 9, 456. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, M.; Wang, X.; Ying, W.; Hu, X.; Dai, P.; Meng, F.; Shi, L.; Sun, Y.; Yao, N.; et al. Tild-CRISPR Allows for Efficient and Precise Gene Knockin in Mouse and Human Cells. Dev. Cell 2018, 45, 526–536.e5. [Google Scholar] [CrossRef]
- Ge, Z.; Zheng, L.; Zhao, Y.; Jiang, J.; Zhang, E.J.; Liu, T.; Gu, H.; Qu, L. Engineered xCas9 and SpCas9-NG variants broaden PAM recognition sites to generate mutations in Arabidopsis plants. Plant Biotechnol. J. 2019, 17, 1865–1867. [Google Scholar] [CrossRef]
- Xue, Z.; Wu, M.; Wen, K.; Ren, M.; Long, L.; Zhang, X.; Gao, G. CRISPR/Cas9 Mediates Efficient Conditional Mutagenesis in Drosophila. G3 Genes|Genomes|Genetics 2014, 4, 2167–2173. [Google Scholar] [CrossRef]
- Batzir, N.A.; Tovin, A.; Hendel, A. Therapeutic Genome Editing and its Potential Enhancement through CRISPR Guide RNA and Cas9 Modifications. Pediatr. Endocrinol. Rev. 2017, 14, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, T.S.; Baudrier, L.; Billon, P.; Ciccia, A. CRISPR-based genome editing through the lens of DNA repair. Mol. Cell 2022, 82, 348–388. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M. Genome Editing: A New Approach to Human Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 163–190. [Google Scholar] [CrossRef]
- Carroll, D. Genome Engineering with Targetable Nucleases. Annu. Rev. Biochem. 2014, 83, 409–439. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005, 23, 967–973. [Google Scholar] [CrossRef]
- Guo, X.; Li, X.-J. Targeted genome editing in primate embryos. Cell Res. 2015, 25, 767–768. [Google Scholar] [CrossRef]
- Urnov, F.D.; Miller, J.C.; Lee, Y.-L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef]
- Lombardo, A.L.; Genovese, P.; Beausejour, C.M.; Colleoni, S.; Lee, Y.-L.; Kim, K.A.; Ando, D.; Urnov, F.D.; Galli, C.; Gregory, P.; et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007, 25, 1298–1306. [Google Scholar] [CrossRef]
- Rai, R.; Thrasher, A.J.; Cavazza, A. Gene Editing for the Treatment of Primary Immunodeficiency Diseases. Hum. Gene Ther. 2021, 32, 43–51. [Google Scholar] [CrossRef]
- Hoban, M.D.; Cost, G.J.; Mendel, M.C.; Romero, Z.; Kaufman, M.L.; Joglekar, A.V.; Ho, M.; Lumaquin, D.; Gray, D.; Lill, G.R.; et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 2015, 125, 2597–2604. [Google Scholar] [CrossRef] [PubMed]
- Cromer, M.K.; Camarena, J.; Martin, R.M.; Lesch, B.J.; Vakulskas, C.A.; Bode, N.M.; Kurgan, G.; Collingwood, M.A.; Rettig, G.R.; Behlke, M.A.; et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 2021, 27, 677–687. [Google Scholar] [CrossRef]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.-J.; Mitros, T.; Muñoz, D.P.; Boffelli, D.; et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef]
- Antony, J.S.; Latifi, N.; Haque, A.K.M.A.; Lamsfus-Calle, A.; Daniel-Moreno, A.; Graeter, S.; Baskaran, P.; Weinmann, P.; Mezger, M.; Handgretinger, R.; et al. Gene correction of HBB mutations in CD34+ hematopoietic stem cells using Cas9 mRNA and ssODN donors. Mol. Cell. Pediatr. 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Hoban, M.D.; Lumaquin, D.; Kuo, C.Y.; Romero, Z.; Long, J.; Ho, M.; Young, C.S.; Mojadidi, M.; Fitz-Gibbon, S.; Cooper, A.R.; et al. CRISPR/Cas9-Mediated Correction of the Sickle Mutation in Human CD34+ cells. Mol. Ther. 2016, 24, 1561–1569. [Google Scholar] [CrossRef]
- Pattabhi, S.; Lotti, S.N.; Berger, M.P.; Singh, S.; Lux, C.T.; Jacoby, K.; Lee, C.; Negre, O.; Scharenberg, A.M.; Rawlings, D.J. In Vivo Outcome of Homology-Directed Repair at the HBB Gene in HSC Using Alternative Donor Template Delivery Methods. Mol. Ther. Nucleic Acids 2019, 17, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Romero, Z.; Lomova, A.; Said, S.; Miggelbrink, A.; Kuo, C.Y.; Campo-Fernandez, B.; Hoban, M.D.; Masiuk, K.E.; Clark, D.N.; Long, J.; et al. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019, 27, 1389–1406. [Google Scholar] [CrossRef]
- Lomova, A.; Clark, D.N.; Campo-Fernandez, B.; Flores-Bjurström, C.; Kaufman, M.L.; Fitz-Gibbon, S.; Wang, X.; Miyahira, E.Y.; Brown, D.; DeWitt, M.A.; et al. Improving Gene Editing Outcomes in Human Hematopoietic Stem and Progenitor Cells by Temporal Control of DNA Repair. Stem Cells 2018, 37, 284–294. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, C.; Dever, D.P.; Davis, T.H.; Camarena, J.; Srifa, W.; Zhang, Y.; Paikari, A.; Chang, A.K.; Porteus, M.H.; et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019, 47, 7955–7972. [Google Scholar] [CrossRef] [PubMed]
- Pavel-Dinu, M.; Gardner, C.L.; Nakauchi, Y.; Kawai, T.; Delmonte, O.M.; Palterer, B.; Bosticardo, M.; Pala, F.; Viel, S.; Malech, H.L.; et al. Genetically Corrected RAG2-SCID Human Hematopoietic Stem Cells Restore V(D)J-Recombinase and Rescue Lymphoid Deficiency. bioRxiv 2022. [Google Scholar] [CrossRef]
- Pavel-Dinu, M.; Wiebking, V.; Dejene, B.T.; Srifa, W.; Mantri, S.; Nicolas, C.E.; Lee, C.; Bao, G.; Kildebeck, E.J.; Punjya, N.; et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019, 10, 1634. [Google Scholar] [CrossRef]
- Goodwin, M.; Lee, E.; Lakshmanan, U.; Shipp, S.; Froessl, L.; Barzaghi, F.; Passerini, L.; Narula, M.; Sheikali, A.; Lee, C.M.; et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci. Adv. 2020, 6, eaaz0571. [Google Scholar] [CrossRef]
- Rai, R.; Romito, M.; Rivers, E.; Turchiano, G.; Blattner, G.; Vetharoy, W.; Ladon, D.; Andrieux, G.; Zhang, F.; Zinicola, M.; et al. Targeted gene correction of human hematopoietic stem cells for the treatment of Wiskott - Aldrich Syndrome. Nat. Commun. 2020, 11, 4034. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Long, J.D.; Campo-Fernandez, B.; de Oliveira, S.; Cooper, A.R.; Romero, Z.; Hoban, M.D.; Joglekar, A.V.; Lill, G.R.; Kaufman, M.L.; et al. Site-Specific Gene Editing of Human Hematopoietic Stem Cells for X-Linked Hyper-IgM Syndrome. Cell Rep. 2018, 23, 2606–2616. [Google Scholar] [CrossRef] [PubMed]
- De Ravin, S.S.; Li, L.; Wu, X.; Choi, U.; Allen, C.; Koontz, S.; Lee, J.; Theobald-Whiting, N.; Chu, J.; Garofalo, M.; et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017, 9, eaah3480. [Google Scholar] [CrossRef]
- Milone, M.C.; Odoherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Loza, L.I.M.; Yuen, E.C.; McCray, P.B. Lentiviral Vectors for the Treatment and Prevention of Cystic Fibrosis Lung Disease. Genes 2019, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- De Ravin, S.S.; Wu, X.; Moir, S.; Kardava, L.; Anaya-Obrien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Su, L.; et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra57. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L. Ex vivo gene transfer and correction for cell-based therapies. Nat. Rev. Genet. 2011, 12, 301–315. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.; Pike-Overzet, K.; Chatters, S.J.; De Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Bartholomae, C.C.; Paruzynski, A.; Arens, A.; Kim, S.; Yu, S.S.; Hong, Y.; Joo, C.-W.; Yoon, N.-K.; Rhim, J.-W.; et al. Retroviral Gene Therapy for X-linked Chronic Granulomatous Disease: Results from Phase I/II Trial. Mol. Ther. 2011, 19, 2092–2101. [Google Scholar] [CrossRef]
- Moratto, D.; Giliani, S.; Bonfim, C.; Mazzolari, E.; Fischer, A.; Ochs, H.D.; Cant, A.J.; Thrasher, A.J.; Cowan, M.J.; Albert, M.H.; et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: An international collaborative study. Blood 2011, 118, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Candotti, F.; Shaw, K.L.; Muul, L.; Carbonaro, D.; Sokolic, R.; Choi, C.; Schurman, S.; Garabedian, E.; Kesserwan, C.; Jagadeesh, G.J.; et al. Gene therapy for adenosine deaminase–deficient severe combined immune deficiency: Clinical comparison of retroviral vectors and treatment plans. Blood 2012, 120, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Nota Informativa Importante—Strimvelis®. Available online: https://www.aifa.gov.it/en/-/nota-informativa-importante-strimvelis- (accessed on 12 September 2022).
- Treatment of SCID Due to ADA Deficiency With Autologous Transplantation of Cord Blood or Hematopoietic CD 34+ Cells After Addition of a Normal Human ADA cDNA by the EFS-ADA Lentiviral Vector—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02022696?term=lentiviral&cond=Ada-Scid&draw=2&rank=2 (accessed on 28 February 2023).
- Gene Therapy for Wiskott-Aldrich Syndrome (WAS)—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01347242?term=lentiviral&cond=WAS&draw=2&rank=3 (accessed on 28 February 2023).
- Ferrua, F.; Cicalese, M.P.; Galimberti, S.; Giannelli, S.; Dionisio, F.; Barzaghi, F.; Migliavacca, M.; Bernardo, M.E.; Calbi, V.; Assanelli, A.A.; et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: Interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019, 6, e239–e253. [Google Scholar] [CrossRef] [PubMed]
- Cowan, M.J.; Yu, J.; Facchino, J.; Fraser-Browne, C.; Sanford, U.; Kawahara, M.; Dara, J.; Long-Boyle, J.; Oh, J.; Chan, W.; et al. Lentiviral Gene Therapy for Artemis-Deficient SCID. N. Engl. J. Med. 2022, 387, 2344–2355. [Google Scholar] [CrossRef]
- Phase I/II Clinical Trial Stem Cell Gene Therapy in RAG1-Deficient SCID—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04797260 (accessed on 28 February 2023).
- Lentiviral Gene Therapy for X-linked Severe Combined Immunodeficiency—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03601286 (accessed on 28 February 2023).
- Perez, L.G.; van Eggermond, M.; van Roon, L.; Vloemans, S.A.; Cordes, M.; Schambach, A.; Rothe, M.; Berghuis, D.; Lagresle-Peyrou, C.; Cavazzana, M.; et al. Successful Preclinical Development of Gene Therapy for Recombinase-Activating Gene-1-Deficient SCID. Mol. Ther. Methods Clin. Dev. 2020, 17, 666–682. [Google Scholar] [CrossRef]
- Autologous Gene Therapy for Artemis-Deficient SCID—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03538899?term=lentiviral&cond=artemis+scid&draw=2&rank=2 (accessed on 28 February 2023).
- Mamcarz, E.; Zhou, S.; Lockey, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Ma, Z.; Condori, J.; Dowdy, J.; Triplett, B.; et al. Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef]
- Kohn, D.B.; Booth, C.; Shaw, K.L.; Xu-Bayford, J.; Garabedian, E.; Trevisan, V.; Carbonaro-Sarracino, D.A.; Soni, K.; Terrazas, D.; Snell, K.; et al. Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency. N. Engl. J. Med. 2021, 384, 2002–2013. [Google Scholar] [CrossRef]
- Marktel, S.; Scaramuzza, S.; Cicalese, M.P.; Giglio, F.; Galimberti, S.; Lidonnici, M.R.; Calbi, V.; Assanelli, A.; Bernardo, M.E.; Rossi, C.; et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat. Med. 2019, 25, 234–241. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N. Safety and Efficacy of Gene-Based Therapeutics for Inherited Disorders; Springer: Berlin/Heidelberg, Germany, 2017; pp. 1–220. [Google Scholar] [CrossRef]
- Yu, N.; Yang, J.; Mishina, Y.; Giannobile, W. Genome Editing: A New Horizon for Oral and Craniofacial Research. J. Dent. Res. 2018, 98, 36–45. [Google Scholar] [CrossRef]
- Xu, X.; Hulshoff, M.S.; Tan, X.; Zeisberg, M.; Zeisberg, E.M. CRISPR/Cas Derivatives as Novel Gene Modulating Tools: Possibilities and In Vivo Applications. Int. J. Mol. Sci. 2020, 21, 3038. [Google Scholar] [CrossRef]
- Mak, A.N.-S.; Bradley, P.; Bogdanove, A.J.; Stoddard, B.L. TAL effectors: Function, structure, engineering and applications. Curr. Opin. Struct. Biol. 2013, 23, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Bak, R.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.; Rosenberg, M.; Hendel, A. Using Synthetically Engineered Guide RNAs to Enhance CRISPR Genome Editing Systems in Mammalian Cells. Front. Genome Ed. 2021, 2, 617910. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.; Iancu, O.; Jacobi, A.M.; McNeill, M.S.; Turk, R.; Rettig, G.R.; Amit, I.; Tovin-Recht, A.; Yakhini, Z.; Behlke, M.A.; et al. Increasing CRISPR Efficiency and Measuring Its Specificity in HSPCs Using a Clinically Relevant System. Mol. Ther. Methods Clin. Dev. 2020, 17, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S. The road less traveled: Strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Cas-mediated transgenesis. BMB Rep. 2018, 51, 437–443. [Google Scholar] [CrossRef]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nature 2019, 21, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Azhagiri, M.K.K.; Babu, P.; Venkatesan, V.; Thangavel, S. Homology-directed gene-editing approaches for hematopoietic stem and progenitor cell gene therapy. Stem Cell Res. Ther. 2021, 12, 500. [Google Scholar] [CrossRef]
- Salisbury-Ruf, C.T.; Larochelle, A. Advances and Obstacles in Homology-Mediated Gene Editing of Hematopoietic Stem Cells. J. Clin. Med. 2021, 10, 513. [Google Scholar] [CrossRef] [PubMed]
- Shy, B.R.; Vykunta, V.S.; Ha, A.; Talbot, A.; Roth, T.L.; Nguyen, D.N.; Pfeifer, W.G.; Chen, Y.Y.; Blaeschke, F.; Shifrut, E.; et al. High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat. Biotechnol. 2022, 41, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef]
- McCarty, D.M. Self-complementary AAV Vectors; Advances and Applications. Mol. Ther. 2008, 16, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Mccarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Kan, Y.; Ruis, B.; Lin, S.; Hendrickson, E.A. The Mechanism of Gene Targeting in Human Somatic Cells. PLoS Genet. 2014, 10, e1004251. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Kantor, B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses 2021, 13, 1288. [Google Scholar] [CrossRef]
- Schubert, M.S.; Thommandru, B.; Woodley, J.; Turk, R.; Yan, S.; Kurgan, G.; McNeill, M.S.; Rettig, G.R. Optimized design parameters for CRISPR Cas9 and Cas12a homology-directed repair. Sci. Rep. 2021, 11, 19482. [Google Scholar] [CrossRef]
- Chen, F.; Pruett-Miller, S.M.; Davis, G.D. Gene Editing Using ssODNs with Engineered Endonucleases. Methods Mol. Biol. 2014, 1239, 251–265. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef]
- Richardson, C.; Ray, G.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Tasca, F.; Wang, Q.; Gonçalves, M.A. Adenoviral Vectors Meet Gene Editing: A Rising Partnership for the Genomic Engineering of Human Stem Cells and Their Progeny. Cells 2020, 9, 953. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Lumbreras, S.; Hernandez-Alcoceba, R. High-Capacity Adenoviral Vectors: Expanding the Scope of Gene Therapy. Int. J. Mol. Sci. 2020, 21, 3643. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Psatha, N.; Gil, S.; Wang, H.; Papayannopoulou, T.; Lieber, A. HDAd5/35++ Adenovirus Vector Expressing Anti-CRISPR Peptides Decreases CRISPR/Cas9 Toxicity in Human Hematopoietic Stem Cells. Mol. Ther.-Methods Clin. Dev. 2018, 9, 390–401. [Google Scholar] [CrossRef]
- Maggio, I.; Gonçalves, M.A. Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol. 2015, 33, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Porteus, M.H. Answered and Unanswered Questions in Early-Stage Viral Vector Transduction Biology and Innate Primary Cell Toxicity for Ex-Vivo Gene Editing. Front. Immunol. 2021, 12, 660302. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Chen, X.; Gonçalves, M.A.F.V. Engineered Viruses as Genome Editing Devices. Mol. Ther. 2016, 24, 447–457. [Google Scholar] [CrossRef]
- Earley, L.F.; Conatser, L.; Lue, V.; Dobbins, M.A.L.; Li, C.; Hirsch, M.L.; Samulski, R.J. Adeno-Associated Virus Serotype-Specific Inverted Terminal Repeat Sequence Role in Vector Transgene Expression. Hum. Gene Ther. 2020, 31, 151–162. [Google Scholar] [CrossRef]
- Bijlani, S.; Pang, K.M.; Sivanandam, V.; Singh, A.; Chatterjee, S. The Role of Recombinant AAV in Precise Genome Editing. Front. Genome Ed. 2022, 3, 799722. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, H.-I.; Li, J.; Sun, L.; Zhang, J.; Xiao, X. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003, 10, 2105–2111. [Google Scholar] [CrossRef]
- Allen, D.; Knop, O.; Itkowitz, B.; Iancu, O.; Beider, K.; Lee, Y.N.; Nagler, A.; Somech, R.; Hendel, A. CRISPR-Cas9 RAG2 Correction Via Coding Sequence Replacement to Preserve Endogenous Gene Regulation and Locus Structure; Research Square: Durham, NC, USA, 2023. [Google Scholar] [CrossRef]
- Bak, R.O.; Dever, D.P.; Porteus, M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018, 13, 358–376. [Google Scholar] [CrossRef]
- Brault, J.; Liu, T.; Liu, S.; Lawson, A.; Choi, U.; Kozhushko, N.; Bzhilyanskaya, V.; Pavel-Dinu, M.; Meis, R.J.; Eckhaus, M.A.; et al. CRISPR-Cas9-AAV versus lentivector transduction for genome modification of X-linked severe combined immunodeficiency hematopoietic stem cells. Front. Immunol. 2023, 13, 1067417. [Google Scholar] [CrossRef]
- De Ravin, S.S.; Reik, A.; Liu, P.-Q.; Li, L.; Wu, X.; Su, L.; Raley, C.; Theobald, N.; Choi, U.; Song, A.H.; et al. Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat. Biotechnol. 2016, 34, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.L.; Pavel-Dinu, M.; Choi, U.; Brault, J.; Liu, T.; Koontz, S.; Li, L.; Theobald, N.; Lee, J.; Bello, E.A.; et al. Correction of X-CGD patient HSPCs by targeted CYBB cDNA insertion using CRISPR/Cas9 with 53BP1 inhibition for enhanced homology-directed repair. Gene Ther. 2021, 28, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Merling, R.K.; Kuhns, U.B.; Sweeney, C.L.; Wu, X.; Burkett, S.; Chu, J.; Lee, J.; Koontz, S.; Di Pasquale, G.; Afione, S.A.; et al. Gene-edited pseudogene resurrection corrects p47phox-deficient chronic granulomatous disease. Blood Adv. 2016, 1, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Pavani, G.; Laurent, M.; Fabiano, A.; Cantelli, E.; Sakkal, A.; Corre, G.; Lenting, P.J.; Concordet, J.-P.; Toueille, M.; Miccio, A.; et al. Ex vivo editing of human hematopoietic stem cells for erythroid expression of therapeutic proteins. Nat. Commun. 2020, 11, 3778. [Google Scholar] [CrossRef]
- Gray, D.H.; Villegas, I.; Long, J.; Santos, J.; Keir, A.; Abele, A.; Kuo, C.Y.; Kohn, D.B. Optimizing Integration and Expression of Transgenic Bruton’s Tyrosine Kinase for CRISPR-Cas9-Mediated Gene Editing of X-Linked Agammaglobulinemia. CRISPR J. 2021, 4, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Brault, J.; Liu, T.Q.; Bello, E.A.; Liu, S.; Sweeney, C.L.; Meis, R.J.; Koontz, S.; Corsino, C.; Choi, U.; Vayssiere, G.; et al. CRISPR-targeted MAGT1 insertion restores XMEN patient hematopoietic stem cells and lymphocytes. Blood 2021, 138, 2768–2780. [Google Scholar] [CrossRef]
- Gardner, C.L.; Pavel-Dinu, M.; Dobbs, K.; Bosticardo, M.; Reardon, P.K.; Lack, J.; DeRavin, S.S.; Le, K.; Bello, E.; Pala, F.; et al. Gene Editing Rescues In vitro T Cell Development of RAG2-Deficient Induced Pluripotent Stem Cells in an Artificial Thymic Organoid System. J. Clin. Immunol. 2021, 41, 852–862. [Google Scholar] [CrossRef]
- Iancu, O.; Allen, D.; Knop, O.; Zehavi, Y.; Breier, D.; Arbiv, A.; Lev, A.; Lee, Y.N.; Beider, K.; Nagler, A.; et al. Multiplex HDR for disease and correction modeling of SCID by CRISPR genome editing in human HSPCs. Mol. Ther.-Nucleic Acids 2022, 31, 105–121. [Google Scholar] [CrossRef]
- Miyazaki, K.; Miyazaki, M. The Interplay Between Chromatin Architecture and Lineage-Specific Transcription Factors and the Regulation of Rag Gene Expression. Front. Immunol. 2021, 12, 659761. [Google Scholar] [CrossRef]
- Allen, D.; Weiss, L.E.; Saguy, A.; Rosenberg, M.; Iancu, O.; Matalon, O.; Lee, C.; Beider, K.; Nagler, A.; Shechtman, Y.; et al. High-Throughput Imaging of CRISPR- and Recombinant Adeno-Associated Virus–Induced DNA Damage Response in Human Hematopoietic Stem and Progenitor Cells. CRISPR J. 2022, 5, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://ir.graphitebio.com/press-releases/detail/84/graphite-bio-announces-voluntary-pause-of-phase-12-cedar (accessed on 5 January 2023).
- Chamberlain, K.; Riyad, J.M.; Weber, T. Expressing Transgenes That Exceed the Packaging Capacity of Adeno-Associated Virus Capsids. Hum. Gene Ther. Methods 2016, 27, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Marrone, L.; Marchi, P.M.; Azzouz, M. Circumventing the packaging limit of AAV-mediated gene replacement therapy for neurological disorders. Expert Opin. Biol. Ther. 2022, 22, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Genovese, P.; Schiroli, G.; Escobar, G.; Di Tomaso, T.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, C.; Holmes, M.C.; et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014, 510, 235–240. [Google Scholar] [CrossRef]
- Banasik, M.B.; McCray, P.B. Integrase-defective lentiviral vectors: Progress and applications. Gene Ther. 2009, 17, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Ortinski, P.I.; O’donovan, B.; Dong, X.; Kantor, B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. Methods Clin. Dev. 2017, 5, 153–164. [Google Scholar] [CrossRef]
- Ferrari, S.; Jacob, A.; Beretta, S.; Unali, G.; Albano, L.; Vavassori, V.; Cittaro, D.; Lazarevic, D.; Brombin, C.; Cugnata, F.; et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat. Biotechnol. 2020, 38, 1298–1308. [Google Scholar] [CrossRef]
- Wienert, B.; Cromer, M.K. CRISPR nuclease off-target activity and mitigation strategies. Front. Genome Ed. 2022, 4, 1050507. [Google Scholar] [CrossRef]
- Naseem, A.; Steinberg, Z.; Cavazza, A. Genome editing for primary immunodeficiencies: A therapeutic perspective on Wiskott-Aldrich syndrome. Front. Immunol. 2022, 13, 4553. [Google Scholar] [CrossRef]
- Höijer, I.; Emmanouilidou, A.; Östlund, R.; van Schendel, R.; Bozorgpana, S.; Tijsterman, M.; Feuk, L.; Gyllensten, U.; Hoed, M.D.; Ameur, A. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat. Commun. 2022, 13, 627. [Google Scholar] [CrossRef]
- Amit, I.; Iancu, O.; Levy-Jurgenson, A.; Kurgan, G.; McNeill, M.S.; Rettig, G.R.; Allen, D.; Breier, D.; Ben Haim, N.; Wang, Y.; et al. CRISPECTOR provides accurate estimation of genome editing translocation and off-target activity from comparative NGS data. Nat. Commun. 2021, 12, 3042. [Google Scholar] [CrossRef]
- Lattanzi, A.; Camarena, J.; Lahiri, P.; Segal, H.; Srifa, W.; Vakulskas, C.A.; Frock, R.L.; Kenrick, J.; Lee, C.; Talbott, N.; et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021, 13, eabf2444. [Google Scholar] [CrossRef]
- Boutin, J.; Rosier, J.; Cappellen, D.; Prat, F.; Toutain, J.; Pennamen, P.; Bouron, J.; Rooryck, C.; Merlio, J.P.; Lamrissi-Garcia, I.; et al. CRISPR-Cas9 globin editing can induce megabase-scale copy-neutral losses of heterozygosity in hematopoietic cells. Nat. Commun. 2021, 12, 4922. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.-Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Adikusuma, F.; Piltz, S.; Corbett, M.A.; Turvey, M.; McColl, S.R.; Helbig, K.J.; Beard, M.R.; Hughes, J.; Pomerantz, R.T.; Thomas, P.Q. Large deletions induced by Cas9 cleavage. Nature 2018, 560, E8–E9. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Turchiano, G.; Andrieux, G.; Klermund, J.; Blattner, G.; Pennucci, V.; el Gaz, M.; Monaco, G.; Poddar, S.; Mussolino, C.; Cornu, T.I.; et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell 2021, 28, 1136–1147.e5. [Google Scholar] [CrossRef]
- Nahmad, A.D.; Reuveni, E.; Goldschmidt, E.; Tenne, T.; Liberman, M.; Horovitz-Fried, M.; Khosravi, R.; Kobo, H.; Reinstein, E.; Madi, A.; et al. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat. Biotechnol. 2022, 40, 1807–1813. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2014, 33, 187–197. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.; Fuller, C.K.; Donohoue, P.D.; Jones, B.N.; Thompson, M.S.; Carter, M.M.; Gradia, S.; Vidal, B.; Garner, E.; Slorach, E.M.; et al. Mapping the genomic landscape of CRISPR–Cas9 cleavage. Nat. Methods 2017, 14, 600–606. [Google Scholar] [CrossRef]
- Ferrari, S.; Jacob, A.; Cesana, D.; Laugel, M.; Beretta, S.; Varesi, A.; Unali, G.; Conti, A.; Canarutto, D.; Albano, L.; et al. Choice of template delivery mitigates the genotoxic risk and adverse impact of editing in human hematopoietic stem cells. Cell Stem Cell 2022, 29, 1428–1444.e9. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.; Cappellen, D.; Rosier, J.; Amintas, S.; Dabernat, S.; Bedel, A.; Moreau-Gaudry, F. ON-Target Adverse Events of CRISPR-Cas9 Nuclease: More Chaotic than Expected. CRISPR J. 2022, 5, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. Int. J. Mol. Sci. 2020, 21, 6461. [Google Scholar] [CrossRef]
- Ferrari, S.; Vavassori, V.; Canarutto, D.; Jacob, A.; Castiello, M.C.; Javed, A.O.; Genovese, P. Gene Editing of Hematopoietic Stem Cells: Hopes and Hurdles Toward Clinical Translation. Front. Genome Ed. 2021, 3, 618378. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Hacein-Bey-Abina, S. Gene therapy for severe combined immunodeficiencies and beyond. J. Exp. Med. 2020, 217, e20190607. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Hauer, J.; Lim, A.; Picard, C.; Wang, G.P.; Berry, C.C.; Martinache, C.; Rieux-Laucat, F.; Latour, S.; Belohradsky, B.H.; et al. Efficacy of Gene Therapy for X-Linked Severe Combined Immunodeficiency. N. Engl. J. Med. 2010, 363, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Clarke, E.L.; Connell, J.; Six, E.; Kadry, N.; Abbas, A.; Hwang, Y.; Everett, J.K.; Hofstaedter, C.; Marsh, R.; Armant, M.; et al. T cell dynamics and response of the microbiota after gene therapy to treat X-linked severe combined immunodeficiency. Genome Med. 2018, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, R.; Yang, D.R.; Puck, J.M.; Huie, M.L.; Jiang, C.-K.; Kurlandsky, L.E. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat. Genet. 1996, 13, 290–295. [Google Scholar] [CrossRef]
- Stephan, V.; Wahn, V.; Le Deist, F.; Dirksen, U.; Bröker, B.; Müller-Fleckenstein, I.; Horneff, G.; Schroten, H.; Fischer, A.; de Saint Basile, G. Atypical X-Linked Severe Combined Immunodeficiency Due to Possible Spontaneous Reversion of the Genetic Defect in T Cells. N. Engl. J. Med. 1996, 335, 1563–1567. [Google Scholar] [CrossRef]
- Speckmann, C.; Pannicke, U.; Wiech, E.; Schwarz, K.; Fisch, P.; Friedrich, W.; Niehues, T.; Gilmour, K.; Buiting, K.; Schlesier, M.; et al. Clinical and immunologic consequences of a somatic reversion in a patient with X-linked severe combined immunodeficiency. Blood 2008, 112, 4090–4097. [Google Scholar] [CrossRef]
- Dvorak, C.C.; Long-Boyle, J.; Dara, J.; Melton, A.; Shimano, K.A.; Huang, J.N.; Puck, J.M.; Dorsey, M.J.; Facchino, J.; Chang, C.K.; et al. Low Exposure Busulfan Conditioning to Achieve Sufficient Multilineage Chimerism in Patients with Severe Combined Immunodeficiency. Biol. Blood Marrow Transplant. 2019, 25, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Canny, M.D.; Moatti, N.; Wan, L.C.K.; Fradet-Turcotte, A.; Krasner, D.; Mateos-Gomez, P.A.; Zimmermann, M.; Orthwein, A.; Juang, Y.-C.; Zhang, W.; et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR–Cas9 genome-editing efficiency. Nat. Biotechnol. 2017, 36, 95–102. [Google Scholar] [CrossRef]
- Bischoff, N.; Wimberger, S.; Maresca, M.; Brakebusch, C. Improving Precise CRISPR Genome Editing by Small Molecules: Is there a Magic Potion? Cells 2020, 9, 1318. [Google Scholar] [CrossRef] [PubMed]
- Schiroli, G.; Ferrari, S.; Conway, A.; Jacob, A.; Capo, V.; Albano, L.; Plati, T.; Castiello, M.C.; Sanvito, F.; Gennery, A.R.; et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017, 9, eaan0820. [Google Scholar] [CrossRef] [PubMed]
- Wimberger, S.; Akrap, N.; Firth, M.; Brengdahl, J.; Engberg, S.; Schwinn, M.K.; Slater, M.R.; Lundin, A.; Hsieh, P.P.; Li, S.; et al. Simultaneous inhibition of DNA-PK and Polϴ improves integration efficiency and precision of genome editing. bioRxiv 2022. [Google Scholar] [CrossRef]
- Belan, O.; Sebald, M.; Adamowicz, M.; Anand, R.; Vancevska, A.; Neves, J.; Grinkevich, V.; Hewitt, G.; Segura-Bayona, S.; Bellelli, R.; et al. POLQ seals post-replicative ssDNA gaps to maintain genome stability in BRCA-deficient cancer cells. Mol. Cell 2022, 82, 4664–4680.e9. [Google Scholar] [CrossRef]
- Shin, J.J.; Schröder, M.S.; Caiado, F.; Wyman, S.K.; Bray, N.L.; Bordi, M.; DeWitt, M.A.; Vu, J.T.; Kim, W.-T.; Hockemeyer, D.; et al. Controlled Cycling and Quiescence Enables Efficient HDR in Engraftment-Enriched Adult Hematopoietic Stem and Progenitor Cells. Cell Rep. 2020, 32, 108093. [Google Scholar] [CrossRef]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegué, E. Hematopoietic Stem Cell Quiescence Promotes Error-Prone DNA Repair and Mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef]
- Piras, F.; Kajaste-Rudnitski, A. Antiviral immunity and nucleic acid sensing in haematopoietic stem cell gene engineering. Gene Ther. 2020, 28, 16–28. [Google Scholar] [CrossRef]
- Piras, F.; Riba, M.; Petrillo, C.; Lazarevic, D.; Cuccovillo, I.; Bartolaccini, S.; Stupka, E.; Gentner, B.; Cittaro, D.; Naldini, L.; et al. Lentiviral vectors escape innate sensing but trigger p53 in human hematopoietic stem and progenitor cells. EMBO Mol. Med. 2017, 9, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Schiroli, G.; Conti, A.; Ferrari, S.; della Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 2019, 24, 551–565.e8. [Google Scholar] [CrossRef] [PubMed]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A Distinctive DNA Damage Response in Human Hematopoietic Stem Cells Reveals an Apoptosis-Independent Role for p53 in Self-Renewal. Cell Stem Cell 2010, 7, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Frati, G.; Miccio, A. Genome Editing for β-Hemoglobinopathies: Advances and Challenges. J. Clin. Med. 2021, 10, 482. [Google Scholar] [CrossRef]
- Zonari, E.; Desantis, G.; Petrillo, C.; Boccalatte, F.E.; Lidonnici, M.R.; Kajaste-Rudnitski, A.; Aiuti, A.; Ferrari, G.; Naldini, L.; Gentner, B. Efficient Ex Vivo Engineering and Expansion of Highly Purified Human Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy. Stem Cell Rep. 2017, 8, 977–990. [Google Scholar] [CrossRef]
- Glimm, H.; Oh, I.H.; Eaves, C.J. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G(2)/M transit and do not reenter G(0). Blood 2000, 96, 4185–4193. [Google Scholar] [CrossRef]
- Kallinikou, K.; Afonso, F.D.A.; Blundell, M.; Ings, S.J.; Watts, M.J.; Thrasher, A.; Linch, D.C.; Bonnet, D.; Yong, K.L. Engraftment defect of cytokine-cultured adult human mobilized CD34(+) cells is related to reduced adhesion to bone marrow niche elements. Br. J. Haematol. 2012, 158, 778–787. [Google Scholar] [CrossRef]
- Larochelle, A.; Gillette, J.M.; Desmond, R.; Ichwan, B.; Cantilena, A.; Cerf, A.; Barrett, A.J.; Wayne, A.S.; Lippincott-Schwartz, J.; Dunbar, C.E. Bone marrow homing and engraftment of human hematopoietic stem and progenitor cells is mediated by a polarized membrane domain. Blood 2012, 119, 1848–1855. [Google Scholar] [CrossRef]
- Poletto, E.; Colella, P.; Vera, L.N.P.; Khan, S.; Tomatsu, S.; Baldo, G.; Gomez-Ospina, N. Improved engraftment and therapeutic efficacy by human genome-edited hematopoietic stem cells with Busulfan-based myeloablation. Mol. Ther.-Methods Clin. Dev. 2022, 25, 392–409. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, D.; Kalter, N.; Rosenberg, M.; Hendel, A. Homology-Directed-Repair-Based Genome Editing in HSPCs for the Treatment of Inborn Errors of Immunity and Blood Disorders. Pharmaceutics 2023, 15, 1329. https://doi.org/10.3390/pharmaceutics15051329
Allen D, Kalter N, Rosenberg M, Hendel A. Homology-Directed-Repair-Based Genome Editing in HSPCs for the Treatment of Inborn Errors of Immunity and Blood Disorders. Pharmaceutics. 2023; 15(5):1329. https://doi.org/10.3390/pharmaceutics15051329
Chicago/Turabian StyleAllen, Daniel, Nechama Kalter, Michael Rosenberg, and Ayal Hendel. 2023. "Homology-Directed-Repair-Based Genome Editing in HSPCs for the Treatment of Inborn Errors of Immunity and Blood Disorders" Pharmaceutics 15, no. 5: 1329. https://doi.org/10.3390/pharmaceutics15051329
APA StyleAllen, D., Kalter, N., Rosenberg, M., & Hendel, A. (2023). Homology-Directed-Repair-Based Genome Editing in HSPCs for the Treatment of Inborn Errors of Immunity and Blood Disorders. Pharmaceutics, 15(5), 1329. https://doi.org/10.3390/pharmaceutics15051329