
Amorphization of Ethenzamide and Ethenzamide Cocrystals—A Case Study of Single and Binary Systems Forming Low-Melting Eutectic Phases Loaded on/in Silica Gel

 , , , and
, , , and
Abstract
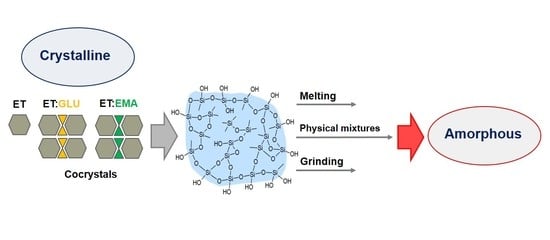
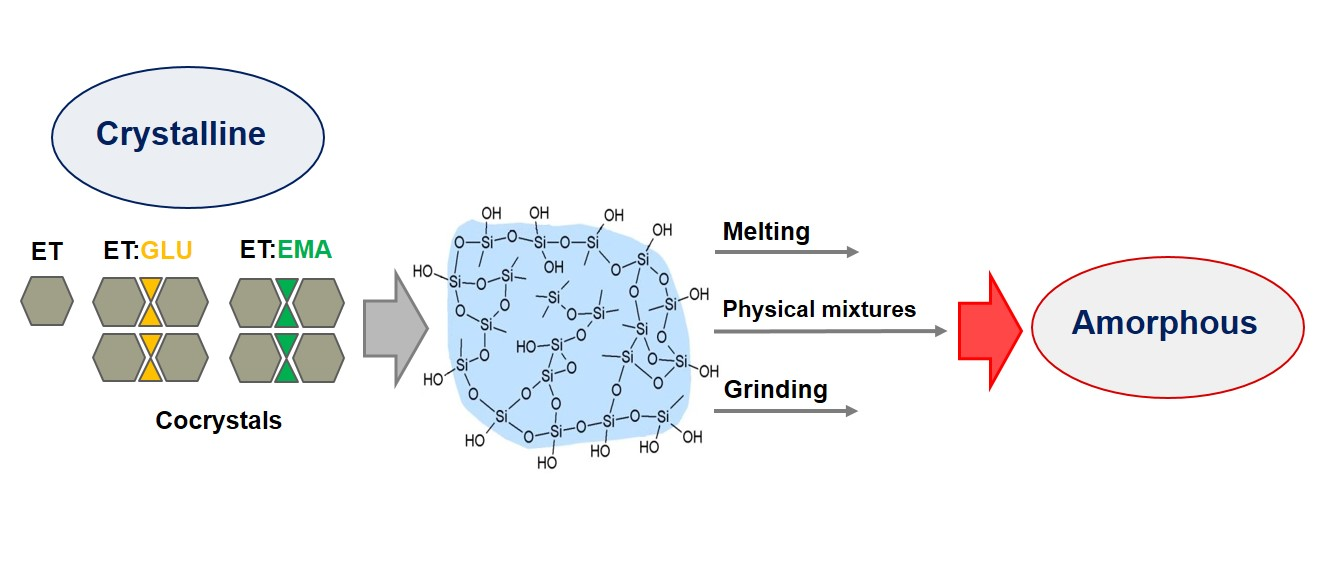{kind=link}
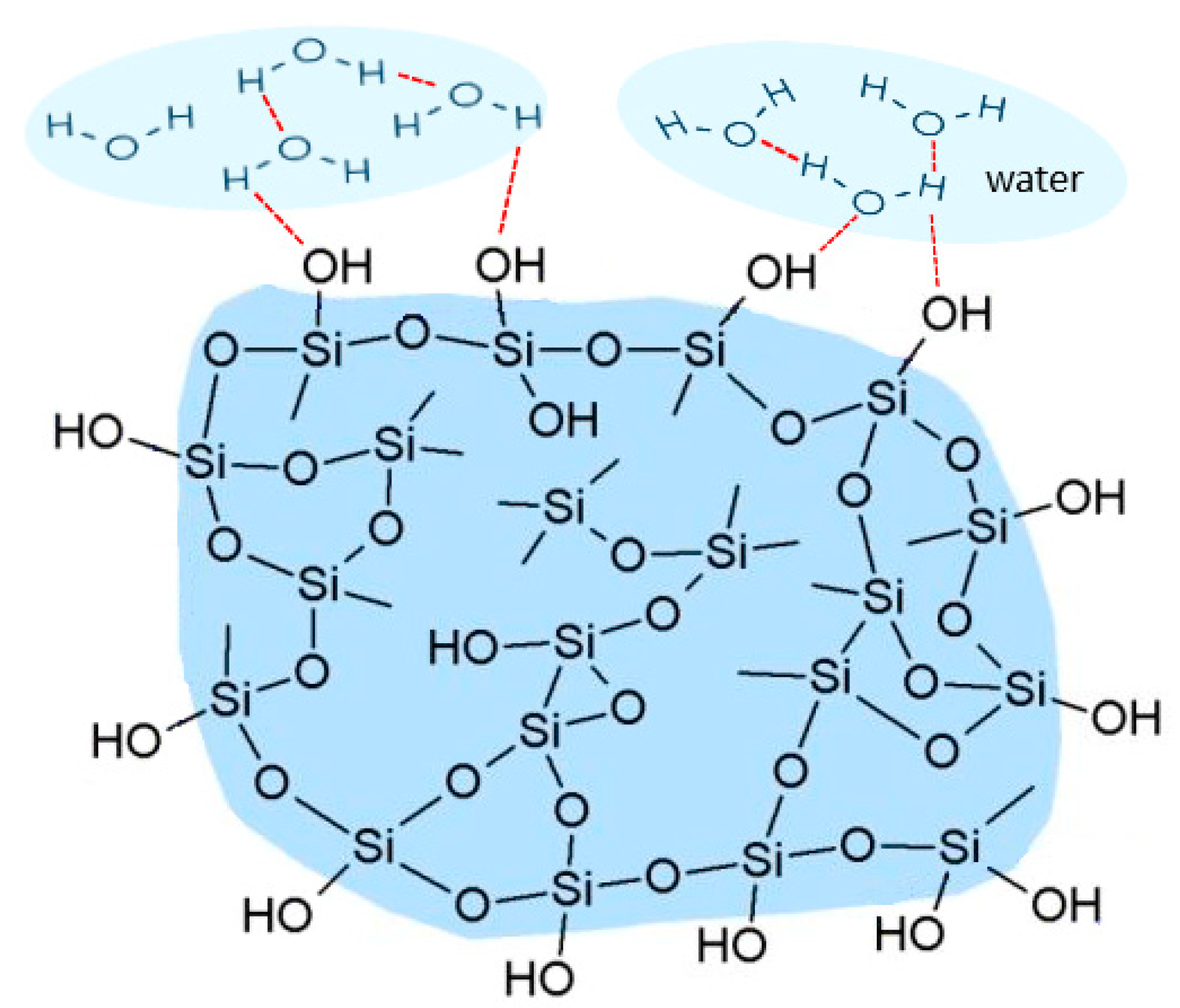{kind=link}
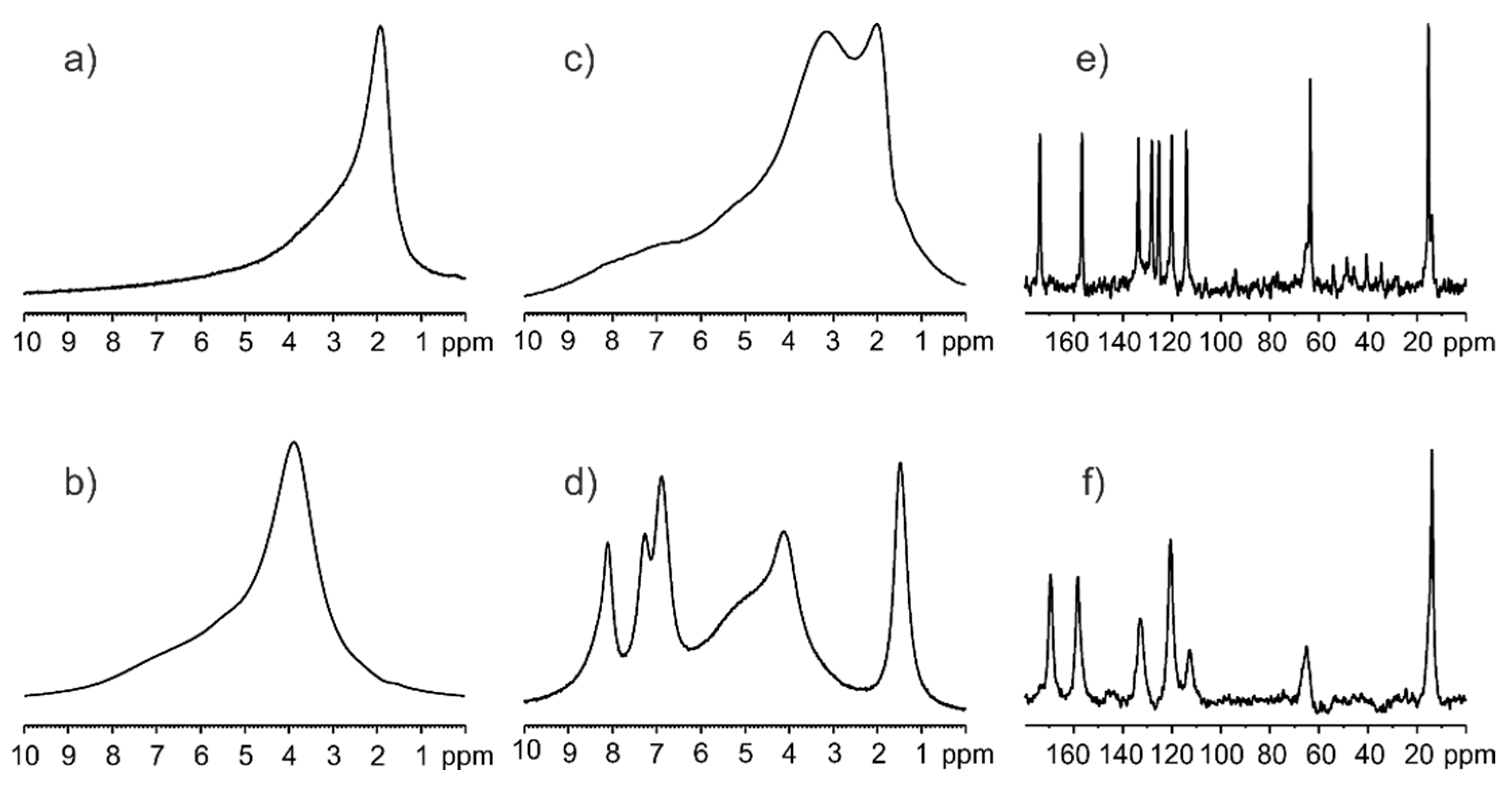{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cocrystals Preparation
2.3. Manual Mixing (MM)
2.4. Thermal Method (TM)
2.5. Ball Milling Method (BM)
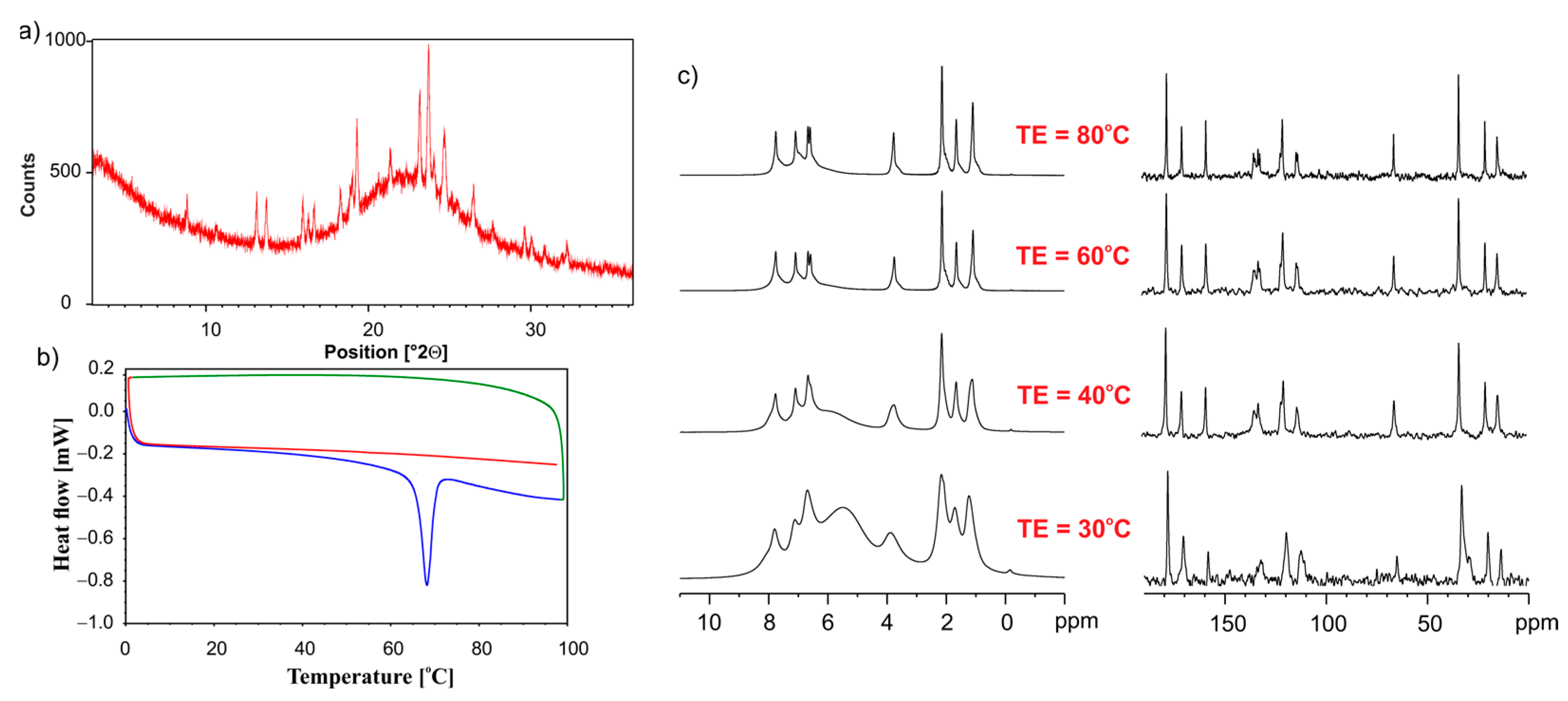
2.6. PXRD Measurements
2.7. Solid-State NMR Experiments
2.8. Differential Scanning Calorimetry
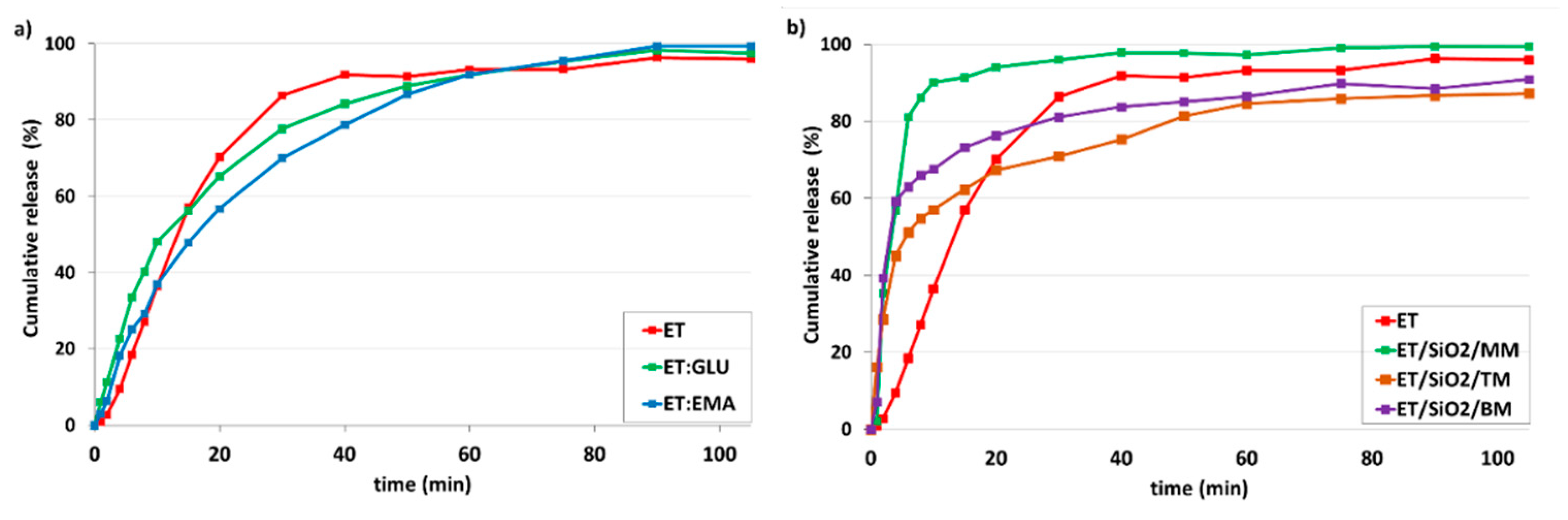
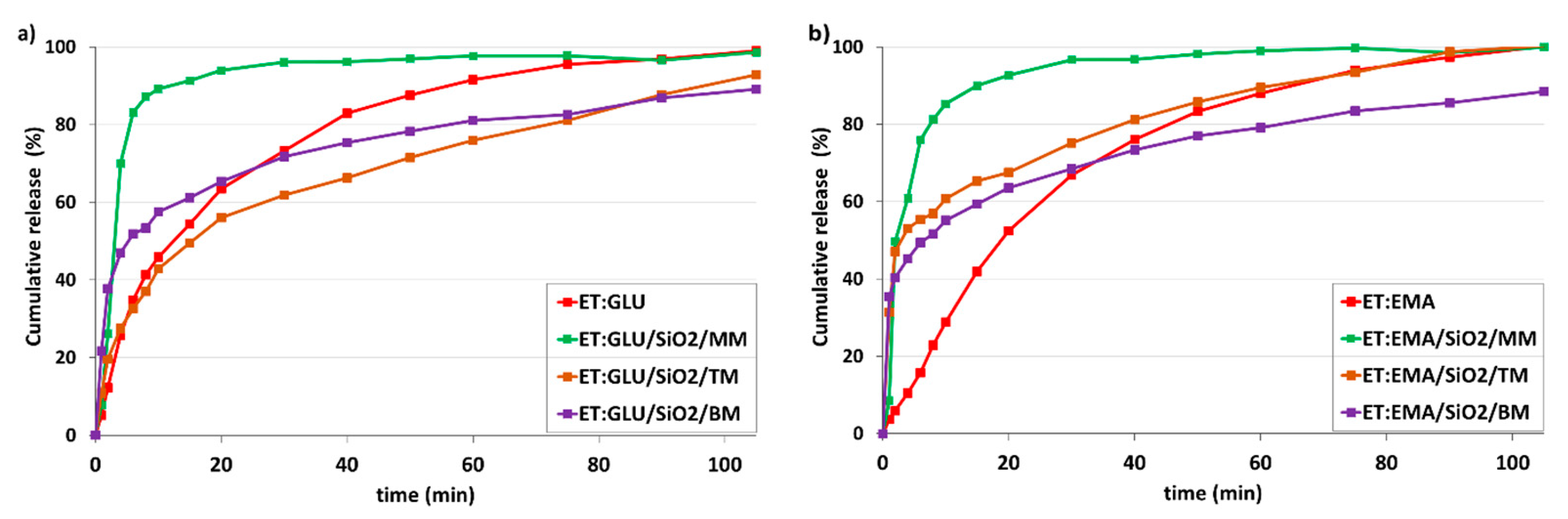
2.9. Dissolution
3. Results
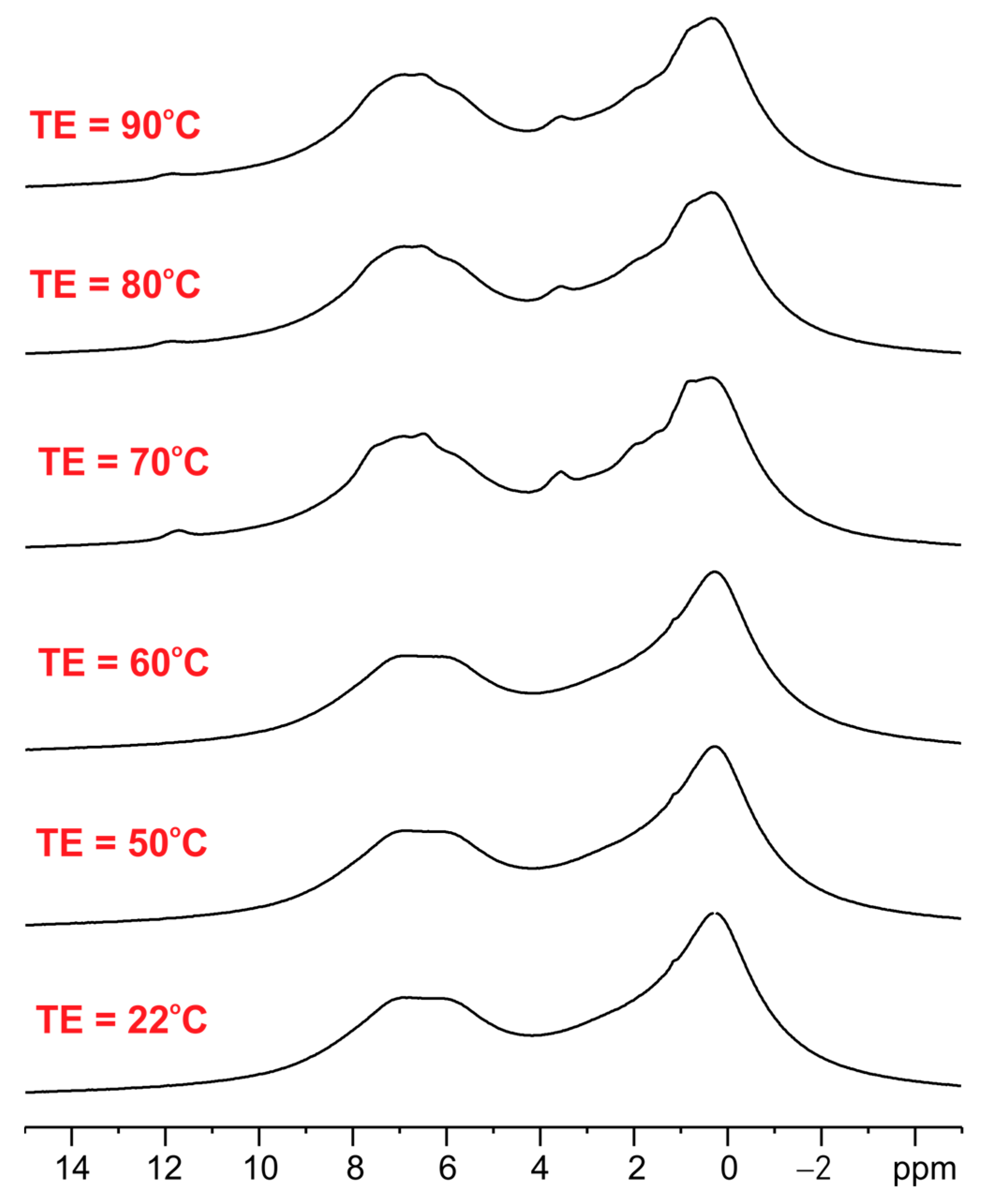
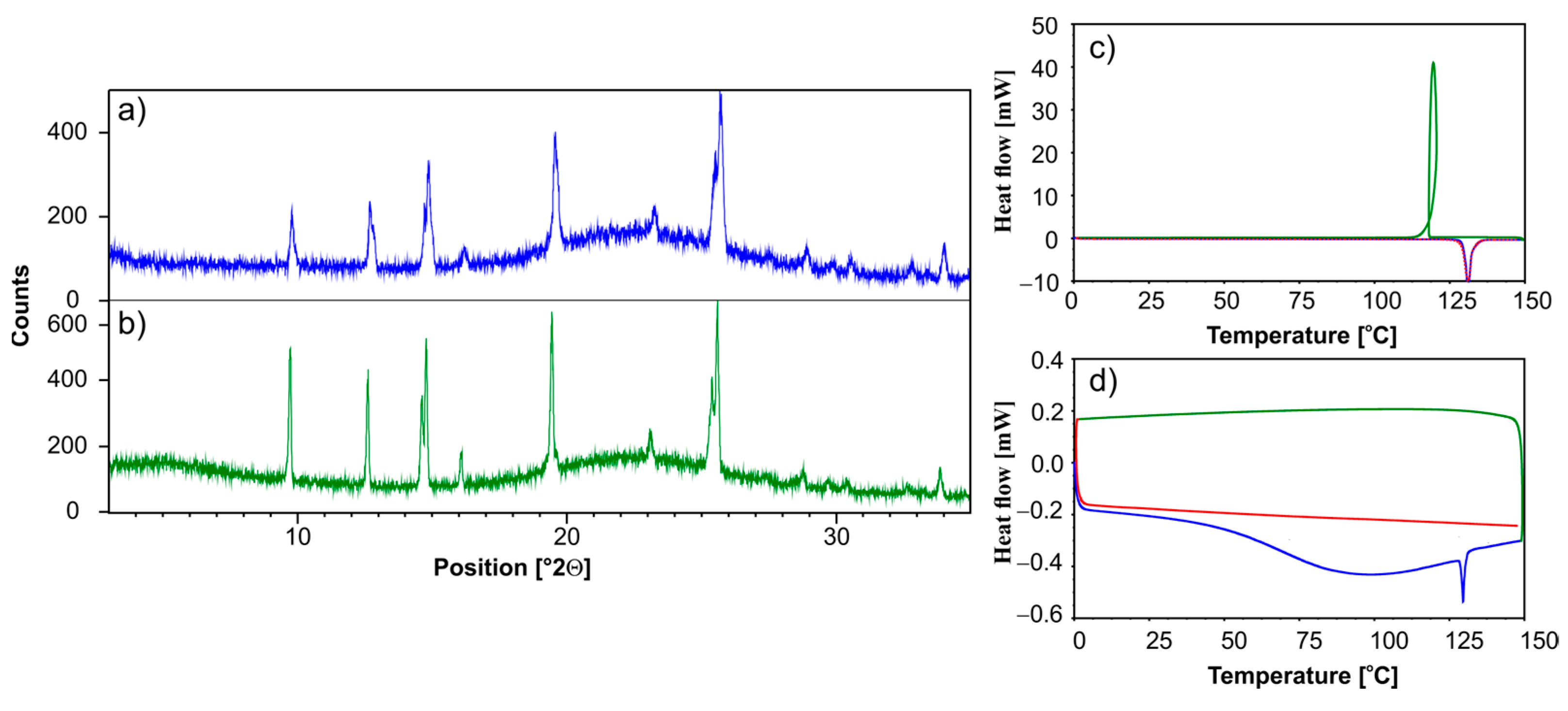
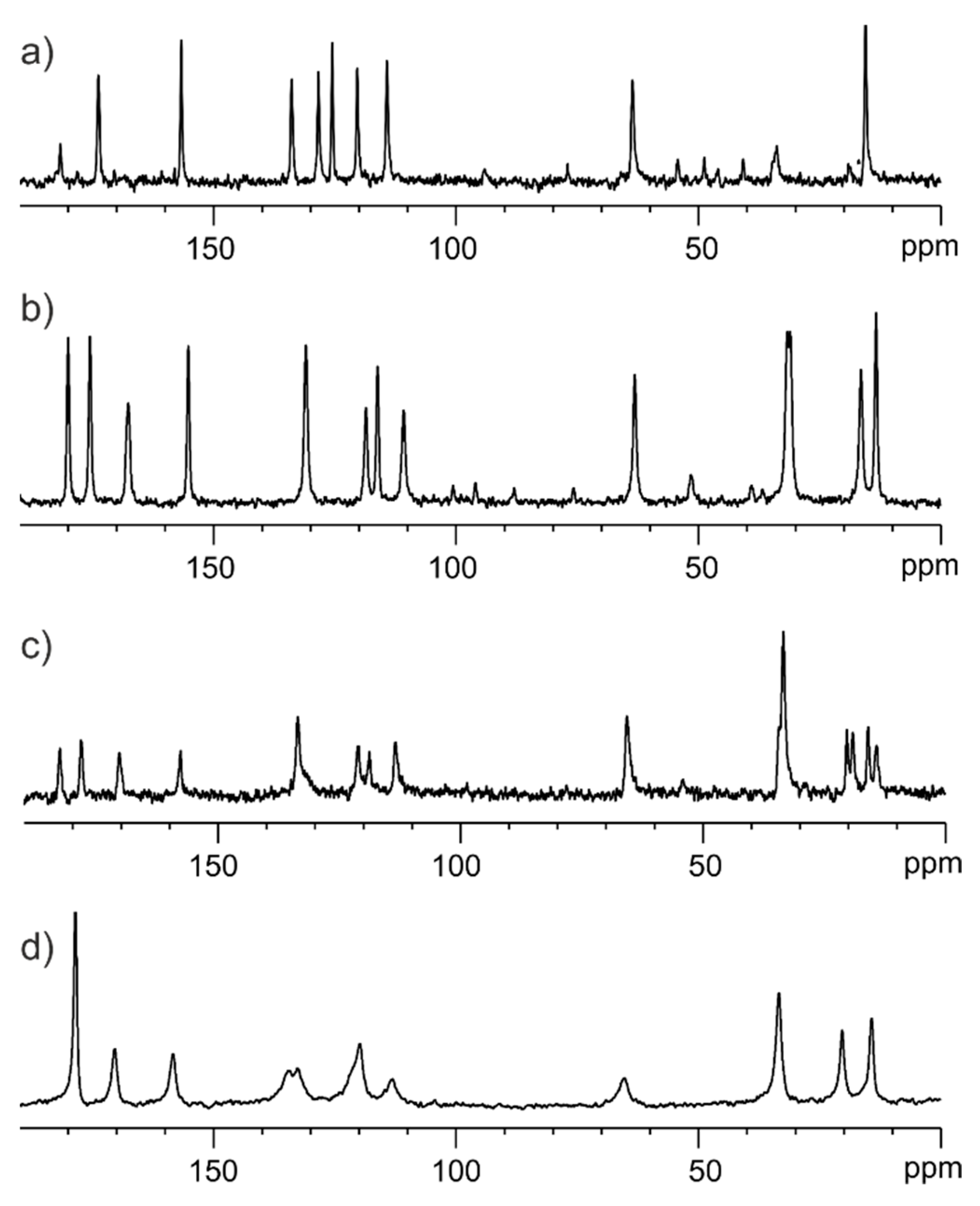
3.1. Solid-State NMR, Differential Scanning Calorimetry (DSC), and Powder X-ray Diffraction (PXRD) Studies of Ethenzamide with Silica
3.1.1. Analysis of Physical Mixture
3.1.2. Analysis of ET/SiO2 Systems Melted or Ground in a Ball Mill
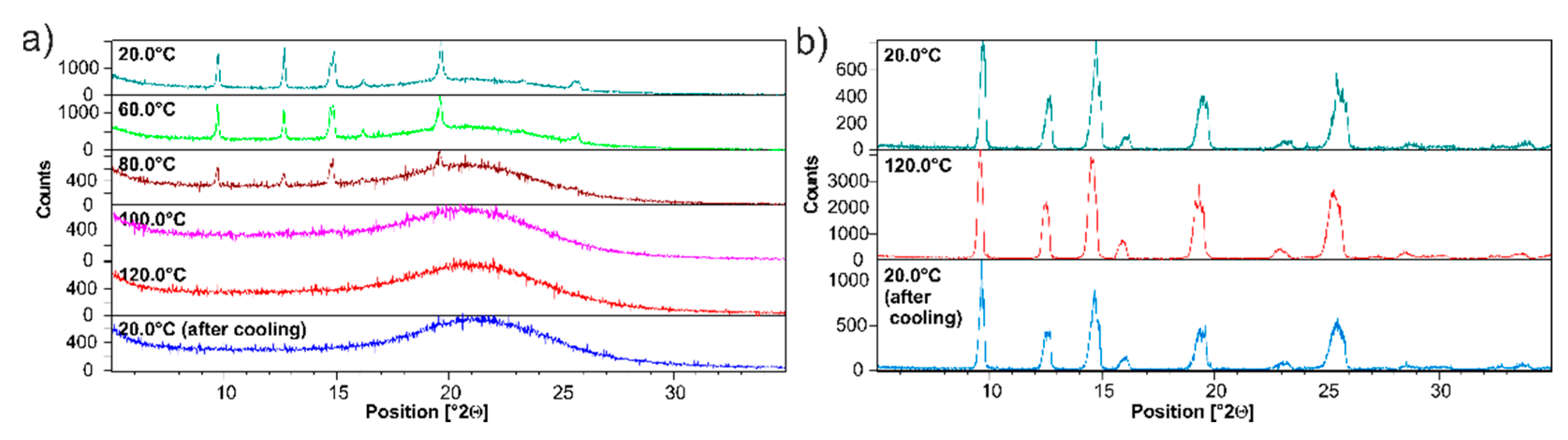
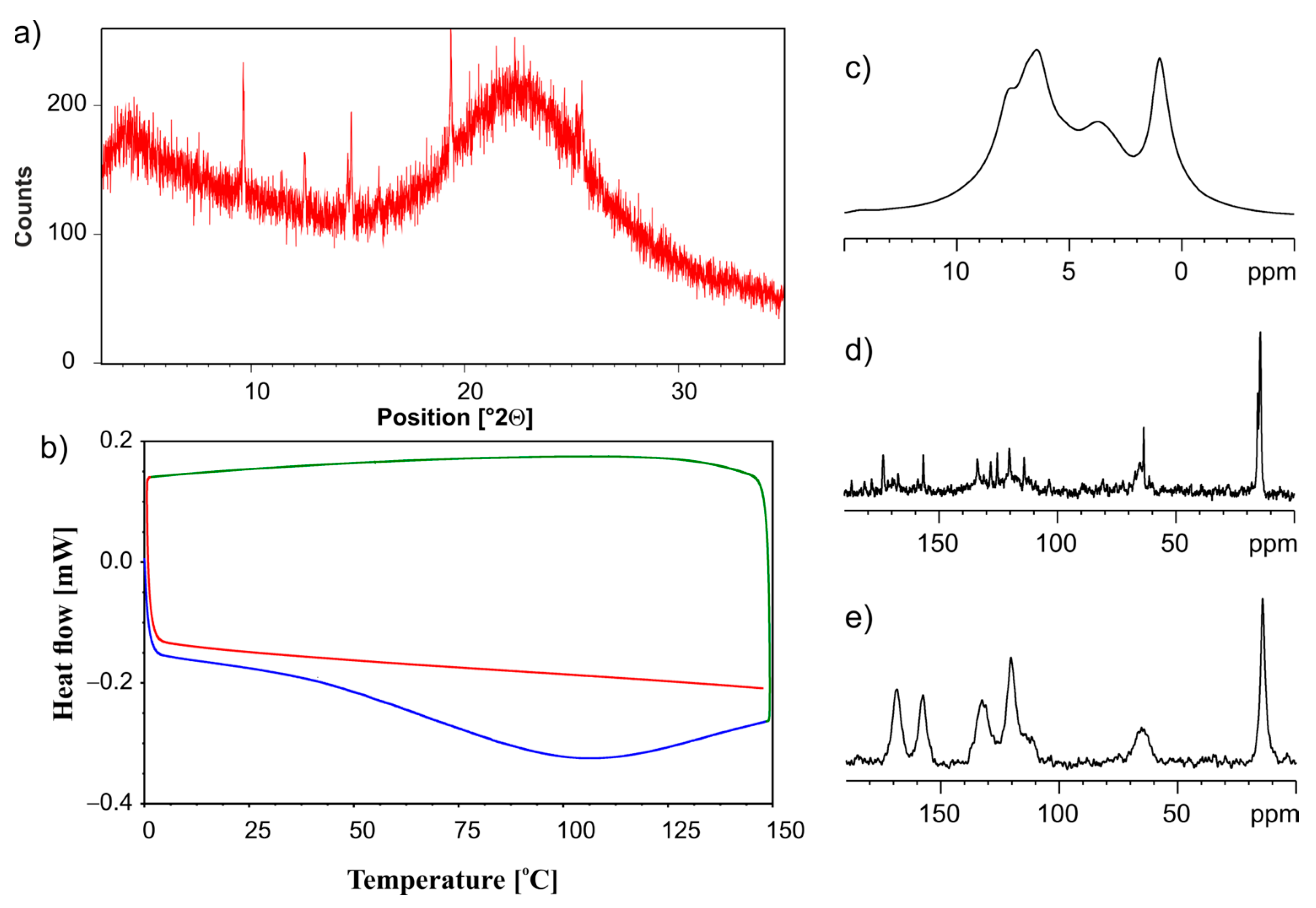
3.2. Silica-Particles-Based Amorphization of ET:EMA and ET:GLU Cocrystals
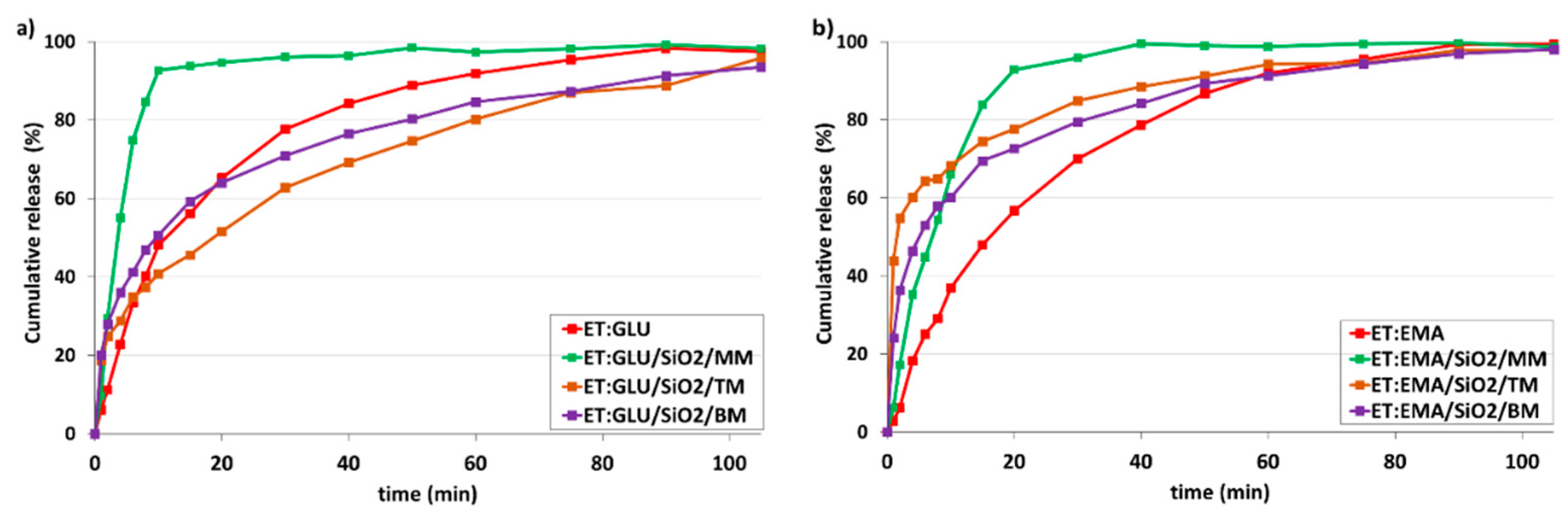
3.3. Dissolution Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation Design for Poorly Water-Soluble Drugs Based on Biopharmaceutics Classification System: Basic Approaches and Practical Applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef]
- Shah, S.; Date, A.; Holm, R. Strategies for the Formulation Development of Poorly Soluble Drugs via Oral Route. In Methods and Principles in Medicinal Chemistry; Bachhav, Y., Ed.; Wiley: Hoboken, NJ, USA, 2019; pp. 49–89. ISBN 978-3-527-34396-6. [Google Scholar]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical Particle Technologies: An Approach to Improve Drug Solubility, Dissolution and Bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Szaniawska, M. Strategies in Poorly Soluble Drug Delivery Systems. Ann. Univ. Mariae Curie-Sklodowska Sect. AA—Chem. 2019, 73, 81. [Google Scholar] [CrossRef]
- Rodriguez-Aller, M.; Guillarme, D.; Veuthey, J.-L.; Gurny, R. Strategies for Formulating and Delivering Poorly Water-Soluble Drugs. J. Drug Deliv. Sci. Technol. 2015, 30, 342–351. [Google Scholar] [CrossRef]
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical Cocrystals: New Solid Phase Modification Approaches for the Formulation of APIs. Pharmaceutics 2018, 10, 18. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Gupta, D.; Bhatia, D.; Dave, V.; Sutariya, V.; Varghese Gupta, S. Salts of Therapeutic Agents: Chemical, Physicochemical, and Biological Considerations. Molecules 2018, 23, 1719. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Salt Formation to Improve Drug Solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Cerreia Vioglio, P.; Chierotti, M.R.; Gobetto, R. Pharmaceutical Aspects of Salt and Cocrystal Forms of APIs and Characterization Challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef] [PubMed]
- van Hoogevest, P.; Liu, X.; Fahr, A. Drug Delivery Strategies for Poorly Water-Soluble Drugs: The Industrial Perspective. Expert Opin. Drug Deliv. 2011, 8, 1481–1500. [Google Scholar] [CrossRef]
- Adepu, S.; Ramakrishna, S. Controlled Drug Delivery Systems: Current Status and Future Directions. Molecules 2021, 26, 5905. [Google Scholar] [CrossRef] [PubMed]
- Stephen, S.; Gorain, B.; Choudhury, H.; Chatterjee, B. Exploring the Role of Mesoporous Silica Nanoparticle in the Development of Novel Drug Delivery Systems. Drug Deliv. Transl. Res. 2022, 12, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.S.; Hassaballah, M.Y.; Abdelqawy, M.A.; Emad-Eldin, M.; Farag, A.K.; Negida, A.; Ghaith, H.; Emam, S.E. An Updated Review of Mesoporous Carbon as a Novel Drug Delivery System. Drug Dev. Ind. Pharm. 2021, 47, 1029–1037. [Google Scholar] [CrossRef]
- Seljak, K.B.; Kocbek, P.; Gašperlin, M. Mesoporous Silica Nanoparticles as Delivery Carriers: An Overview of Drug Loading Techniques. J. Drug Deliv. Sci. Technol. 2020, 59, 101906. [Google Scholar] [CrossRef]
- Moritz, M.; Geszke-Moritz, M. Mesoporous Materials as Elements of Modern Drug Delivery Systems for Anti-Inflammatory Agents: A Review of Recent Achievements. Pharmaceutics 2022, 14, 1542. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, O.N.; Croker, D.M.; Walker, G.M.; Zaworotko, M.J. Pharmaceutical Cocrystals: From Serendipity to Design to Application. Drug Discov. Today 2019, 24, 796–804. [Google Scholar] [CrossRef]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Raheem Thayyil, A.; Juturu, T.; Nayak, S.; Kamath, S. Pharmaceutical Co-Crystallization: Regulatory Aspects, Design, Characterization, and Applications. Adv. Pharm. Bull. 2020, 10, 203–212. [Google Scholar] [CrossRef]
- Port, A.; Almansa, C.; Enrech, R.; Bordas, M.; Plata-Salamán, C.R. Differential Solution Behavior of the New API–API Co-Crystal of Tramadol–Celecoxib (CTC) versus Its Constituents and Their Combination. Cryst. Growth Des. 2019, 19, 3172–3182. [Google Scholar] [CrossRef]
- Sekhon, B.S. Drug-Drug Co-Crystals. DARU J. Pharm. Sci. 2012, 20, 45. [Google Scholar] [CrossRef]
- Shaikh, T.R.; George, C.P.; Bhukya, P.; Shelke, N.; Pawar, K.; Garai, A.; Dandela, R.; Gonnade, R.G.; Nangia, A.K. Novel Crystal Forms of Entresto: A Supramolecular Complex of Trisodium Sacubitril/Valsartan Hemi-Pentahydrate. CrystEngComm 2022, 24, 7387–7393. [Google Scholar] [CrossRef]
- Ngilirabanga, J.B.; Samsodien, H. Pharmaceutical Co-crystal: An Alternative Strategy for Enhanced Physicochemical Properties and Drug Synergy. Nano Sel. 2021, 2, 512–526. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Pharmaceutical Cocrystals: Walking the Talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nanda, A. Pharmaceutical Cocrystals: An Overview. Indian J. Pharm. Sci. 2017, 79, 858–871. [Google Scholar] [CrossRef]
- Kim, D.; Kim, Y.; Tin, Y.-Y.; Soe, M.-T.-P.; Ko, B.; Park, S.; Lee, J. Recent Technologies for Amorphization of Poorly Water-Soluble Drugs. Pharmaceutics 2021, 13, 1318. [Google Scholar] [CrossRef] [PubMed]
- Tambe, S.; Jain, D.; Meruva, S.K.; Rongala, G.; Juluri, A.; Nihalani, G.; Mamidi, H.K.; Nukala, P.K.; Bolla, P.K. Recent Advances in Amorphous Solid Dispersions: Preformulation, Formulation Strategies, Technological Advancements and Characterization. Pharmaceutics 2022, 14, 2203. [Google Scholar] [CrossRef]
- Van den Mooter, G. The Use of Amorphous Solid Dispersions: A Formulation Strategy to Overcome Poor Solubility and Dissolution Rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Solid Dispersion of Poorly Water-soluble Drugs: Early Promises, Subsequent Problems, and Recent Breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.B.; Thipparaboina, R.; Kumar, D.; Shastri, N.R. Co Amorphous Systems: A Product Development Perspective. Int. J. Pharm. 2016, 515, 403–415. [Google Scholar] [CrossRef]
- Han, J.; Wei, Y.; Lu, Y.; Wang, R.; Zhang, J.; Gao, Y.; Qian, S. Co-Amorphous Systems for the Delivery of Poorly Water-Soluble Drugs: Recent Advances and an Update. Expert Opin. Drug Deliv. 2020, 17, 1411–1435. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Grohganz, H.; Löbmann, K.; Rades, T.; Hempel, N.-J. Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, In Vitro and In Vivo Performance, and New Formulation Strategies. Pharmaceutics 2021, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Chieng, N.; Aaltonen, J.; Saville, D.; Rades, T. Physical Characterization and Stability of Amorphous Indomethacin and Ranitidine Hydrochloride Binary Systems Prepared by Mechanical Activation. Eur. J. Pharm. Biopharm. 2009, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, K.; Chotera-Ouda, A.; Bak-Sypien, I.I.; Potrzebowski, M.J. Mesoporous Silica Particles as Drug Delivery Systems—The State of the Art in Loading Methods and the Recent Progress in Analytical Techniques for Monitoring These Processes. Pharmaceutics 2021, 13, 950. [Google Scholar] [CrossRef]
- Maleki, A.; Kettiger, H.; Schoubben, A.; Rosenholm, J.M.; Ambrogi, V.; Hamidi, M. Mesoporous Silica Materials: From Physico-Chemical Properties to Enhanced Dissolution of Poorly Water-Soluble Drugs. J. Control. Release 2017, 262, 329–347. [Google Scholar] [CrossRef]
- Qian, K.K.; Bogner, R.H. Application of Mesoporous Silicon Dioxide and Silicate in Oral Amorphous Drug Delivery Systems. J. Pharm. Sci. 2012, 101, 444–463. [Google Scholar] [CrossRef]
- Trzeciak, K.; Kaźmierski, S.; Wielgus, E.; Potrzebowski, M.J. DiSupLo—New Extremely Easy and Efficient Method for Loading of Active Pharmaceutical Ingredients into the Pores of MCM-41 Mesoporous Silica Particles. Microporous Mesoporous Mater. 2020, 308, 110506. [Google Scholar] [CrossRef]
- Trzeciak, K.; Kaźmierski, S.; Drużbicki, K.; Potrzebowski, M.J. Mapping of Guest Localization in Mesoporous Silica Particles by Solid-State NMR and Ab Initio Modeling: New Insights into Benzoic Acid and p-Fluorobenzoic Acid Embedded in MCM-41 via Ball Milling. J. Phys. Chem. C 2021, 125, 10096–10109. [Google Scholar] [CrossRef]
- Trzeciak, K.; Wielgus, E.; Kaźmierski, S.; Khalaji, M.; Dudek, M.K.; Potrzebowski, M.J. Unexpected Factors Affecting the Kinetics of Guest Molecule Release from Investigation of Binary Chemical Systems Trapped in a Single Void of Mesoporous Silica Particles. ChemPhysChem 2023, 24, e202200884. [Google Scholar] [CrossRef]
- Skorupska, E.; Jeziorna, A.; Paluch, P.; Potrzebowski, M.J. Ibuprofen in Mesopores of Mobil Crystalline Material 41 (MCM-41): A Deeper Understanding. Mol. Pharm. 2014, 11, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Skorupska, E.; Kaźmierski, S.; Potrzebowski, M.J. Solid State NMR Characterization of Ibuprofen:Nicotinamide Cocrystals and New Idea for Controlling Release of Drugs Embedded into Mesoporous Silica Particles. Mol. Pharm. 2017, 14, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Lin, W.; Scholes, P.; Li, M. Investigating the Effects of Loading Factors on the In Vitro Pharmaceutical Performance of Mesoporous Materials as Drug Carriers for Ibuprofen. Materials 2017, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Cha, K.-H.; Hong, S.H.; Abuzar, S.M.; Lee, S.; Ha, E.-S.; Kim, J.-S.; Baek, I.-H.; Kim, M.-S.; Hwang, S.-J. Pharmaceutical Characterization and In Vivo Evaluation of Orlistat Formulations Prepared by the Supercritical Melt-Adsorption Method Using Carbon Dioxide: Effects of Mesoporous Silica Type. Pharmaceutics 2020, 12, 333. [Google Scholar] [CrossRef]
- Azad, M.; Moreno, J.; Davé, R. Stable and Fast-Dissolving Amorphous Drug Composites Preparation via Impregnation of Neusilin® UFL2. J. Pharm. Sci. 2018, 107, 170–182. [Google Scholar] [CrossRef]
- Kovačević, M.; German Ilić, I.; Bolko Seljak, K.; Zvonar Pobirk, A. High-Shear Wet Granulation of SMEDDS Based on Mesoporous Carriers for Improved Carvedilol Solubility. Pharmaceutics 2022, 14, 2077. [Google Scholar] [CrossRef]
- Omachi, Y. Gastroretentive Sustained-Release Tablets Combined with a Solid Self-Micro-Emulsifying Drug Delivery System Adsorbed onto Fujicalin®. AAPS PharmSciTech 2022, 23, 157. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Alotaibi, H.A.; Almehmady, A.M.; Safo, M.K.; El-Say, K.M. Influences of Glimepiride Self-Nanoemulsifying Drug Delivery System Loaded Liquisolid Tablets on the Hypoglycemic Activity and Pancreatic Histopathological Changes in Streptozotocin-Induced Hyperglycemic Rats. Nanomaterials 2022, 12, 3966. [Google Scholar] [CrossRef]
- Skorupska, E.; Jeziorna, A.; Potrzebowski, M.J. Thermal Solvent-Free Method of Loading of Pharmaceutical Cocrystals into the Pores of Silica Particles: A Case of Naproxen/Picolinamide Cocrystal. J. Phys. Chem. C 2016, 120, 13169–13180. [Google Scholar] [CrossRef]
- Galindres, D.M.; Cifuentes, D.; Tinoco, L.E.; Murillo-Acevedo, Y.; Rodrigo, M.M.; Ribeiro, A.C.F.; Esteso, M.A. A Review of the Application of Resorcinarenes and SBA-15 in Drug Delivery. Processes 2022, 10, 684. [Google Scholar] [CrossRef]
- Fathi Vavsari, V.; Mohammadi Ziarani, G.; Badiei, A. The Role of SBA-15 in Drug Delivery. RSC Adv. 2015, 5, 91686–91707. [Google Scholar] [CrossRef]
- Qian, K.K.; Bogner, R.H. Spontaneous Crystalline-to-Amorphous Phase Transformation of Organic or Medicinal Compounds in the Presence of Porous Media, Part 1: Thermodynamics of Spontaneous Amorphization. J. Pharm. Sci. 2011, 100, 2801–2815. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.K.; Suib, S.L.; Bogner, R.H. Spontaneous Crystalline-to-amorphous Phase Transformation of Organic or Medicinal Compounds in the Presence of Porous Media, Part 2: Amorphization Capacity and Mechanisms of Interaction. J. Pharm. Sci. 2011, 100, 4674–4686. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.K.; Wurster, D.E.; Bogner, R.H. Spontaneous Crystalline-to-Amorphous Phase Transformation of Organic or Medicinal Compounds in the Presence of Porous Media, Part 3: Effect of Moisture. Pharm. Res. 2012, 29, 2698–2709. [Google Scholar] [CrossRef]
- Lin, J.H.; Sugiyama, Y.; Awazu, S.; Hanano, M. Physiological Pharmacokinetics of Ethoxybenzamide Based on Biochemical Data Obtainedin Vitro as Well as on Physiological Data. J. Pharmacokinet. Biopharm. 1982, 10, 649–661. [Google Scholar] [CrossRef]
- Kozak, A.; Pindelska, E. Spectroscopic Analysis of the Influence of Various External Factors on Ethenzamide-Glutaric Acid (1:1) Cocrystal Formation. Eur. J. Pharm. Sci. 2019, 133, 59–68. [Google Scholar] [CrossRef]
- Aitipamula, S.; Chow, P.S.; Tan, R.B.H. Conformational and Enantiotropic Polymorphism of a 1:1 Cocrystal Involving Ethenzamide and Ethylmalonic Acid. CrystEngComm 2010, 12, 3691. [Google Scholar] [CrossRef]
- Chu, P.J.; Potrzebowski, M.J.; Scott, A.I.; Gao, Y. Conformational Studies of N-Benzoyl-L-Phenylalanine by Combined Rotation and Multiple-Pulse Spectroscopy Proton Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1990, 112, 881–883. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef]
- Hronský, V. Measurement of Sample Temperatures and Temperature Gradients in Magic-Angle Spinning Nmr. Acta Electrotech. Inform. 2013, 13, 95–98. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Generally Recognized as Safe (GRAS). Available online: https://www.fda.gov/food/food-ingredients-packaging/generally-recognized-safe-gras (accessed on 16 February 2023).
- Gonçalves, M.C. Sol-Gel Silica Nanoparticles in Medicine: A Natural Choice. Design, Synthesis and Products. Molecules 2018, 23, 2021. [Google Scholar] [CrossRef] [PubMed]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trzeciak, K.; Wielgus, E.; Kaźmierski, S.; Pawlak, T.; Potrzebowski, M.J. Amorphization of Ethenzamide and Ethenzamide Cocrystals—A Case Study of Single and Binary Systems Forming Low-Melting Eutectic Phases Loaded on/in Silica Gel. Pharmaceutics 2023, 15, 1234. https://doi.org/10.3390/pharmaceutics15041234
Trzeciak K, Wielgus E, Kaźmierski S, Pawlak T, Potrzebowski MJ. Amorphization of Ethenzamide and Ethenzamide Cocrystals—A Case Study of Single and Binary Systems Forming Low-Melting Eutectic Phases Loaded on/in Silica Gel. Pharmaceutics. 2023; 15(4):1234. https://doi.org/10.3390/pharmaceutics15041234
Chicago/Turabian StyleTrzeciak, Katarzyna, Ewelina Wielgus, Sławomir Kaźmierski, Tomasz Pawlak, and Marek J. Potrzebowski. 2023. "Amorphization of Ethenzamide and Ethenzamide Cocrystals—A Case Study of Single and Binary Systems Forming Low-Melting Eutectic Phases Loaded on/in Silica Gel" Pharmaceutics 15, no. 4: 1234. https://doi.org/10.3390/pharmaceutics15041234
APA StyleTrzeciak, K., Wielgus, E., Kaźmierski, S., Pawlak, T., & Potrzebowski, M. J. (2023). Amorphization of Ethenzamide and Ethenzamide Cocrystals—A Case Study of Single and Binary Systems Forming Low-Melting Eutectic Phases Loaded on/in Silica Gel. Pharmaceutics, 15(4), 1234. https://doi.org/10.3390/pharmaceutics15041234