
Magnetic Solid-Phase Microextraction Protocol Based on Didodecyldimethylammonium Bromide-Functionalized Nanoparticles for the Quantification of Epirubicin in Biological Matrices

 , , , and
, , , and 
Abstract
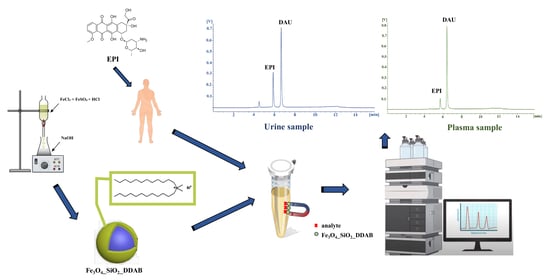
1. Introduction
2. Materials and Methods
2.1. The Chemicals and Reagents
2.2. Chromatographic Conditions
2.3. Stock and Working Standard Solutions
2.4. Calibration and Quality Control Samples
2.5. Method Validation
2.6. Real Samples
3. Results and Discussion
3.1. Optimization of Extraction Procedure
3.1.1. Amount of DDAB
3.1.2. Amount of Adsorbent
3.1.3. Effect of pH
3.1.4. Salting-Out Effect
3.1.5. Extraction and Desorption Time
3.2. Method Validation
3.2.1. Linearity
3.2.2. LOD and LOQ
3.2.3. Selectivity
3.2.4. Inter- and Intra-Day Precision and Accuracy
3.2.5. Stability
3.2.6. Extraction Recovery
3.3. Time–Concentration Profile of EPI in Patient Samples
3.4. Comparison of the Developed MSPME-Based Method with Previous Reports
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Preparation of Magnetic Sorbents
References
- Parmar, S.; Sting, J.C.; Huber-Wechselberger, A.; Kainz, A.; Renner, W.; Langsenlehner, U.; Krippl, P.; Brockmöller, J.; Haschke-Becher, E. Impact of UGT2B7 His268Tyr polymorphism on the outcome of adjuvant epirubicin treatment in breast cancer. Breast Cancer Res. 2011, 13, R57. [Google Scholar] [CrossRef] [PubMed]
- Sayal, K.; Gounaris, I.; Basu, B.; Freeman, S.; Moyle, P.; Hosking, K.; Iddawela, M.; Jimenez-Linan, M.; Abraham, J.; Brenton, J.; et al. Epirubicin, Cisplatin, and Capecitabine for Primary Platinum-Resistant or Platinum-Refractory Epithelial Ovarian Cancer: Results of a Retrospective, Single-Institution Study. Int. J. Gynecol. Cancer 2015, 25, 977. [Google Scholar] [CrossRef] [PubMed]
- Mashhadi, M.; Rahimi, H.; Najafi, B.; Khosravi, A. IEV (Ifosfamide, Epirubicin, VP16) Regimen as Salvage Therapy for Early Relapsed/Refractory non-Hodgkin/Hodgkin Lymphoma. Int. J. Hematol.-Oncol. Stem Cell Res. 2011, 5, 11–15. [Google Scholar]
- Hirai, I.; Tanese, K.; Nakamura, Y.; Ishii, M.; Kawakami, Y.; Funakoshi, T. Combination Cisplatin-Epirubicin-Paclitaxel Therapy for Metastatic Extramammary Paget’s Disease. Oncologist 2019, 24, e394–e396. [Google Scholar] [CrossRef] [PubMed]
- Tarpgaard, L.S.; Qvortrup, C.; Nielsen, S.L.; Stenvang, J.; Detlefsen, S.; Brünner, N.; Pfeiffer, P. New use for old drugs: Epirubicin in colorectal cancer. Acta Oncol. 2021, 60, 954–956. [Google Scholar] [CrossRef]
- Luiz, M.T.; Dutra, J.A.P.; Di Filippo, L.D.; Tavares, A.G., Jr.; Bueno Tofani, L.; Maldonado Marchetti, J.; Chorilli, M. Epirubicin: Biological Properties, Analytical Methods, and Drug Delivery Nanosystems. Crit. Rev. Anal. Chem. 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Ruiz, A.; Nuñez-Torres, R.; Pita, G.; Wildiers, H.; Lambrechts, D.; Hatse, S.; Delombaerde, D.; Van Brussel, T.; Alonso, M.R.; Alvarez, N.; et al. POLRMT as a Novel Susceptibility Gene for Cardiotoxicity in Epirubicin Treatment of Breast Cancer Patients. Pharmaceutics 2021, 13, 1942. [Google Scholar] [CrossRef]
- Beumer, J.H.; Chu, E.; Allegra, C.; Tanigawara, Y.; Milano, G.; Diasio, R.; Kim, T.W.; Mathijssen, R.H.; Zhang, L.; Arnold, D.; et al. Therapeutic Drug Monitoring in Oncology: International Association of Therapeutic Drug Monitoring and Clinical Toxicology Recommendations for 5-Fluorouracil Therapy. Clin. Pharmacol. Ther. 2019, 105, 598–613. [Google Scholar] [CrossRef]
- Li, R.; Dong, L.; Huang, J. Hydrophilic Interaction Chromatographic Determination of Epirubicin in Human Plasma using Solid Phase Extraction for Sample Clean-Up. J. Liq. Chromatogr. Relat. Technol. 2007, 30, 2409–2418. [Google Scholar] [CrossRef]
- Treder, N.; Maliszewska, O.; Olędzka, I.; Kowalski, P.; Miękus, N.; Bączek, T.; Bień, E.; Krawczyk, M.A.; Adamkiewicz-Drożynska, E.; Plenis, A. Development and validation of a high-performance liquid chromatographic method with a fluorescence detector for the analysis of epirubicin in human urine and plasma, and its application in drug monitoring. J. Chromatogr. B 2020, 1136, 121910. [Google Scholar] [CrossRef]
- Alshymaa, A.A.; Górecki, T. Green Approaches to Sample Preparation Based on Extraction Techniques. Molecules 2020, 25, 1719. [Google Scholar] [CrossRef]
- Susanti, I.; Holik, H.A. Review: Application of Magnetic Solid-Phase Extraction (Mspe) in Various Types of Samples. Int. J. Appl. Pharm. 2021, 13, 59–68. [Google Scholar] [CrossRef]
- Heidari, H.; Limouei-Khosrowshahi, B. Magnetic solid phase extraction with carbon-coated Fe3O4 nanoparticles coupled to HPLC-UV for the simultaneous determination of losartan, carvedilol, and amlodipine besylate in plasma samples. J. Chromatogr. B 2019, 1114–1115, 24–30. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, S.; Yang, Y.; Yulan Deng, Y.; Li, D.; Su, P.; Yang, Y. Modification of polydopamine-coated Fe3O4 nanoparticles with multi-walled carbon nanotubes for magnetic-μ-dispersive solid-phase extraction of antiepileptic drugs in biological matrices. Anal. Bioanal. Chem. 2018, 410, 3779–3788. [Google Scholar] [CrossRef]
- Amoli-Diva, M.; Pourghazi, K.; Pourasadollah-Karani, H. Surfactant coated magnetic nanoparticle-based solid-phase extraction coupled with spectrophotometric detection for determination of ultra-trace amounts of indomethacin in biological fluids. Micro Nano Lett. 2015, 10, 135–139. [Google Scholar] [CrossRef]
- Du, J.; Wu, H.; Du, X.; Zhao, Z.; Zhao, X.; Shi, Y.; Guo, X.; Du, L. Magnetic mixed hemimicelle solid-phase extraction based on mixed hemi-/ad-micelle SDS-coated magnetic nanoparticles Fe2-xAlxO3 (x = 0.4) for the fluorimetric determination of carvedilol in biological samples. Can. J. Chem. 2017, 95, 512–519. [Google Scholar] [CrossRef]
- Treder, N.; Roszkowska, A.; Olędzka, I.; Bączek, T.; Plenis, A. Effects of Fe3O4 Magnetic Nanoparticle Functionalization with Ionic Liquids and a Double-Chained Surfactant on the Pretreatment of Plasma Samples during Drug Extraction. Anal. Chem. 2022, 94, 16587–16595. [Google Scholar] [CrossRef]
- European Medicines Agency. Guide to Bioanalytical Method Validation, Committee for Medicinal Products for Human Use and European Medicines Agency. 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guidelinebioanalytical-method-validation_en.pdf (accessed on 23 August 2019).
- US Food Drug Administration. FDA Guidance for Industry: Bioanalytical Method Validation; US Department of Health and Human, Services Food and Drug Administration: Silver Spring, MD, USA, 2013. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 23 August 2019).
- Tafti, F.F.; Masrournia, M. A simple and straightforward combination of surfactant-assisted magnetic dispersive micro-solid-phase extraction and hydride generation procedure to determine arsenic (III) species in environmental, biological, and fruit juice samples. J. Iran. Chem. Soc. 2022, 19, 2383–2394. [Google Scholar] [CrossRef]
- Fard, M.R.; Pourghobadi, Z. The Spectrophotometric Determination of Nystatin in Real Samples Using Solid Phase Extraction Based on Sodium Dodecyl Sulphate-Coated Magnetite Nanoparticles. Anal. Bioanal. Chem. Res. 2018, 5, 249–259. [Google Scholar] [CrossRef]
- Abbasi, L.; Faraji, M.; Bahmaie, M. Extracting trace amount of terbinafine hydrochloride in biological fluids and wastewater samples using solid phase-extraction based on magnetic nanoparticles followed by HPLC-UV analysis. Asia-Pac. J. Chem. Eng. 2014, 9, 826–833. [Google Scholar] [CrossRef]
- Zomoorodi, Z.B.; Masrournia, M.; Abedi, M.R. An in situ modification sorbent for magnetic dispersive micro solid-phase extraction of anti-inflammatory drugs in the human urine sample before their determination with high-performance liquid chromatography. Chem. Pap. 2021, 75, 5813–5824. [Google Scholar] [CrossRef]
- Mushtaq, M.M.W.; Kanwal, F.; Imran, M.; Ameen, N.; Batool, S.; Batool, A.; Bashir, S.; Abbas, S.M.; Rehman, A.; Riaz, S. Synthesis of Surfactant-Coated Cobalt Ferrite Nanoparticles for Adsorptive Removal of Acid Blue 45 Dye. Matter. Res. Express 2018, 5, 11–14. [Google Scholar] [CrossRef]
- Griffin, L.R.; Browning, K.L.; Truscott, C.L.; Clifton, L.A.; Webster, J.; Clarke, S.M. A comparison of didodecyldimethylammonium bromide adsorbed at mica/water and silica/water interfaces using neutron reflection. J.Colloid Interface Sci. 2016, 478, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Azmiyawati, C.; Yamin, F.A.; Darmawan, A.; Suyati, L. Synthesis of magnetite@SILICA-CTA in a cetyl trimethyl ammonium bromide (CTAB) concentration variations for fenol adsorption. AIP Conf. Proc. 2020, 2237, 020006. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, H.; Xiao, D.; Chuong, P.H.; He, J.; He, H. Mixed hemimicelles solid-phase extraction of cephalosporins in biological samples with ionic liquid-coated magnetic graphene oxide nanoparticles coupled with high-performance liquid chromatographic analysis. J. Chromatogr. A 2016, 1454, 1–8. [Google Scholar] [CrossRef]
- Li, R.; Dong, L.; Huang, J. Ultra performance liquid chromatography-tandem mass spectrometry for the determination of epirubicin in human plasma. Anal. Chim. Acta 2005, 546, 167–173. [Google Scholar] [CrossRef]
- Bermingham, S.; O’Connor, R.; Regan, F.; McMahon, G.P. Simultaneous determination of anthracyclines and taxanes in human serum using online sample extraction coupled to high performance liquid chromatography with UV detection. J. Sep. Sci. 2010, 33, 1571–1579. [Google Scholar] [CrossRef]
- Maudens, K.E.; Stove, C.P.; Cocquyt, V.F.J.; Denys, H.; Lambert, W.E. Development and validation of a liquid chromatographic method for the simultaneous determination of four anthracyclines and their respective 13-S-dihydro metabolites in plasma and saliva. J. Chromatogr. B Anal. Tech. Biomed. Life Sci. 2009, 877, 3907–3915. [Google Scholar] [CrossRef]
- Dodde, W.I.W.J.; Maring, G.; Hendriks, G.; Wachters, F.M.; Groen, H.J.M.; De Vries, E.G.E.; Uges, D.R.A. Determination of epirubicin and its metabolite epirubicinol in saliva and plasma by HPLC. Ther. Drug Monit. 2003, 25, 433–440. [Google Scholar] [CrossRef]
- Sottani, C.; Leoni, E.; Porro, B.; Montagna, B.; Amatu, A.; Sottotetti, F.; Quaretti, P.; Poggi, G.; Minoia, C. Validation of an LC-MS/MS method for the determination of epirubicin in human serum of patients undergoing Drug Eluting Microsphere-transarterial Chemoembolization (DEM-TACE). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3543–3548. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrices | QCs | Concentration Added [µg/mL] | Precision (RSD (%), n = 6) | Accuracy (%, n = 6) | ||
|---|---|---|---|---|---|---|
| Intra-Day | Inter-Day | Intra-Day | Inter-Day | |||
| Plasma | Low | 0.1 | 6.26 | 7.78 | 103 | 95 |
| Medium | 0.5 | 4.46 | 3.45 | 101 | 102 | |
| High | 1.0 | 2.93 | 1.97 | 98 | 99 | |
| Urine | Low | 0.5 | 6.61 | 8.57 | 90 | 90 |
| Medium | 3.0 | 4.87 | 5.31 | 97 | 104 | |
| High | 7.5 | 3.12 | 5.02 | 99 | 101 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Treder, N.; Szuszczewicz, N.; Roszkowska, A.; Olędzka, I.; Bączek, T.; Bień, E.; Krawczyk, M.A.; Plenis, A. Magnetic Solid-Phase Microextraction Protocol Based on Didodecyldimethylammonium Bromide-Functionalized Nanoparticles for the Quantification of Epirubicin in Biological Matrices. Pharmaceutics 2023, 15, 1227. https://doi.org/10.3390/pharmaceutics15041227
Treder N, Szuszczewicz N, Roszkowska A, Olędzka I, Bączek T, Bień E, Krawczyk MA, Plenis A. Magnetic Solid-Phase Microextraction Protocol Based on Didodecyldimethylammonium Bromide-Functionalized Nanoparticles for the Quantification of Epirubicin in Biological Matrices. Pharmaceutics. 2023; 15(4):1227. https://doi.org/10.3390/pharmaceutics15041227
Chicago/Turabian StyleTreder, Natalia, Natalia Szuszczewicz, Anna Roszkowska, Ilona Olędzka, Tomasz Bączek, Ewa Bień, Małgorzata Anna Krawczyk, and Alina Plenis. 2023. "Magnetic Solid-Phase Microextraction Protocol Based on Didodecyldimethylammonium Bromide-Functionalized Nanoparticles for the Quantification of Epirubicin in Biological Matrices" Pharmaceutics 15, no. 4: 1227. https://doi.org/10.3390/pharmaceutics15041227
APA StyleTreder, N., Szuszczewicz, N., Roszkowska, A., Olędzka, I., Bączek, T., Bień, E., Krawczyk, M. A., & Plenis, A. (2023). Magnetic Solid-Phase Microextraction Protocol Based on Didodecyldimethylammonium Bromide-Functionalized Nanoparticles for the Quantification of Epirubicin in Biological Matrices. Pharmaceutics, 15(4), 1227. https://doi.org/10.3390/pharmaceutics15041227