
The Current Status, Challenges, and Future Potential of Therapeutic Vaccination in Glioblastoma


Abstract
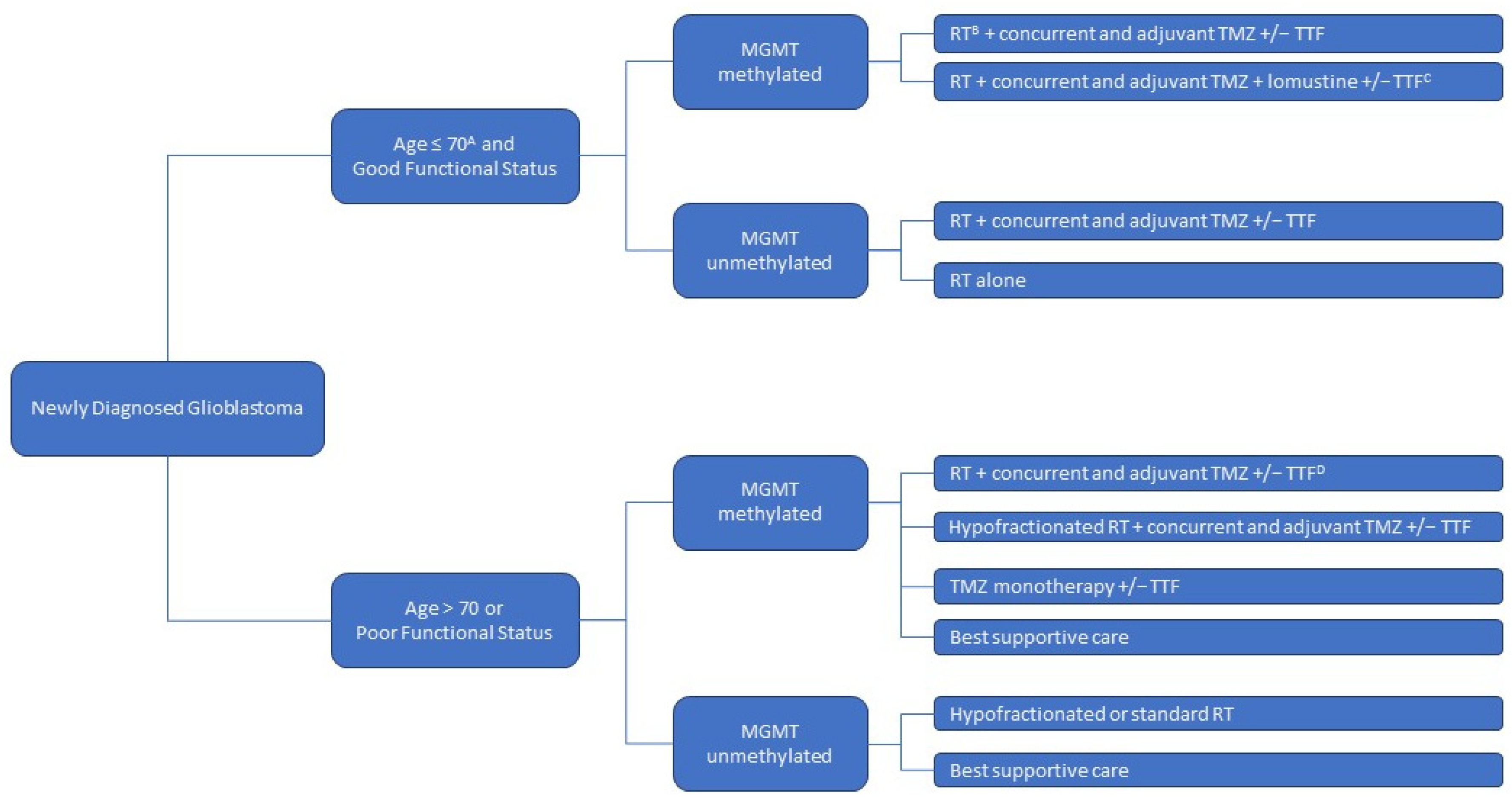
1. Background
2. Immunosuppressive State and Immune-Targeted Therapies
3. Therapeutic Vaccines
4. Delivery
5. Challenges
6. Adjuvant Therapies
7. Logistical Considerations
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, A.K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro-Oncology 2022, 24, v1–v95. [Google Scholar] [CrossRef]
- Roelcke, U.; Schwyzer, L.; Zeitlberger, A.M.; Hundsberger, T. Symptom burden in glioblastoma-a prospective pilot study from diagnosis to first progression. Oncology 2022, 101, 145–152. [Google Scholar] [CrossRef]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Neth, B.J.; Carabenciov, I.D.; Ruff, M.W.; Johnson, D.R. Temporal Trends in Glioblastoma Survival: Progress then Plateau. Neurologist 2021, 27, 119–124. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Mutter, N.; Stupp, R. Temozolomide: A milestone in neuro-oncology and beyond? Expert Rev. Anticancer Ther. 2006, 6, 1187–1204. [Google Scholar] [CrossRef]
- Fabian, D.; Eibl, M.D.P.G.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.-D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA–09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef]
- Malmström, A.; Grønberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, E.M.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.-Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.I.; Iwama, T.; Park, D.M. The evidence of glioblastoma heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef]
- Wang, L.; Jung, J.; Babikir, H.; Shamardani, K.; Jain, S.; Feng, X.; Gupta, N.; Rosi, S.; Chang, S.; Raleigh, D.; et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Cancer 2022, 3, 1534–1552. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 22, 351. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2017, 20, 184–191. [Google Scholar] [CrossRef]
- Liu, H.-L.; Hua, M.-Y.; Chen, P.-Y.; Chu, P.-C.; Pan, C.-H.; Yang, H.-W.; Huang, C.-Y.; Wang, J.-J.; Yen, T.-C.; Wei, K.-C. Blood-Brain Barrier Disruption with Focused Ultrasound Enhances Delivery of Chemotherapeutic Drugs for Glioblastoma Treatment. Radiology 2010, 255, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma: Figure 1. Neuro-Oncology 2015, 17, vii9–vii14. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Waziri, A. Glioblastoma-Derived Mechanisms of Systemic Immunosuppression. Neurosurg. Clin. North Am. 2010, 21, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Himes, B.T.; Peterson, E.T.; de Mooij, T.; Garcia, L.M.C.; Jung, M.-Y.; Uhm, S.; Yan, D.; Tyson, J.; Jin-Lee, H.J.; Parney, D.; et al. The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro-Oncology 2020, 22, 967–978. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Desland, F.A.; Hormigo, A. The CNS and the Brain Tumor Microenvironment: Implications for Glioblastoma Immunotherapy. Int. J. Mol. Sci. 2020, 21, 7358. [Google Scholar] [CrossRef]
- Jackson, C.; Ruzevick, J.; Phallen, J.; Belcaid, Z.; Lim, M. Challenges in Immunotherapy Presented by the Glioblastoma Multiforme Microenvironment. J. Immunol. Res. 2011, 2011, 732413. [Google Scholar] [CrossRef]
- Bornschlegl, S.; Gustafson, M.P.; Delivanis, A.D.; Ryder, M.; Liu, M.C.; Vasmatzis, G.; Hallemeier, C.L.; Park, S.S.; Roberts, L.R.; Parney, I.F.; et al. Categorisation of patients based on immune profiles: A new approach to identifying candidates for response to checkpoint inhibitors. Clin. Transl. Immunol. 2021, 10, e1267. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic immune suppression in glioblastoma: The interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro-Oncology 2010, 12, 631–644. [Google Scholar] [CrossRef]
- Rodrigues, J.C.; Gonzalez, G.C.; Zhang, L.; Ibrahim, G.; Kelly, J.J.; Gustafson, M.P.; Lin, Y.; Dietz, A.; Forsyth, P.A.; Yong, V.W.; et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro-Oncology 2009, 12, 351–365. [Google Scholar] [CrossRef]
- Sener, U.; Ruff, M.W.; Campian, J.L. Immunotherapy in Glioblastoma: Current Approaches and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7046. [Google Scholar] [CrossRef]
- Wilcox, J.A.; Ramakrishna, R.; Magge, R. Immunotherapy in glioblastoma. World Neurosurg. 2018, 116, 518–528. [Google Scholar] [CrossRef]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef]
- van Putten, E.H.P.; Kleijn, A.; Van Beusechem, V.W.; Noske, D.; Lamers, C.H.; De Goede, A.L.; Idema, S.; Hoefnagel, D.; Kloezeman, J.J.; Fueyo, J.; et al. Convection Enhanced Delivery of the Oncolytic Adenovirus Delta24-RGD in Patients with Recurrent GBM: A Phase I Clinical Trial Including Correlative Studies. Clin. Cancer Res. 2022, 28, 1572–1585. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A Review of Cancer Immunotherapy: From the Past, to the Present, to the Future. Curr. Oncol. 2020, 27 (Suppl. S2), S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Roth, P.; Preusser, M.; Wick, W.; Reardon, D.A.; Platten, M.; Sampson, J.H. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat. Rev. Neurol. 2017, 13, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar]
- De Jager, R.; Guinan, E.; Lamm, D.; Khanna, O.; Brosman, S.; De Kernion, J.; Williams, R.; Richardson, C.; Muenz, L.; Reitsma, D.; et al. Long-term complete remission in bladder carcinoma in situ with intravesical TICE bacillus Calmette Guerin. Overview analysis of six phase II clinical trials. Urology 1991, 38, 507–513. [Google Scholar] [CrossRef]
- Sfakianos, J.P.; Salome, B.; Daza, J.; Farkas, A.; Bhardwaj, N.; Horowitz, A. Bacillus Calmette-Guerin (BCG): Its fight against pathogens and cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 39, 121–129. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Chesney, J.A.; Ribas, A.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined with Pembrolizumab for Advanced Melanoma. J. Clin. Oncol. 2023, 41, 528–540. [Google Scholar] [CrossRef]
- Schijns, V.E.J.C.; Pretto, C.; Strik, A.M.; Gloudemans-Rijkers, R.; Devillers, L.; Pierre, D.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.-T.; et al. Therapeutic Immunization against Glioblastoma. Int. J. Mol. Sci. 2018, 19, 2540. [Google Scholar] [CrossRef]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 1–22. [Google Scholar] [CrossRef]
- Frederico, S.C.; Hancock, J.C.; Brettschneider, E.E.S.; Ratnam, N.M.; Gilbert, M.R.; Terabe, M. Making a Cold Tumor Hot: The Role of Vaccines in the Treatment of Glioblastoma. Front. Oncol. 2021, 11, 672508. [Google Scholar] [CrossRef]
- Liu, W.; Tang, H.; Li, L.; Wang, X.; Yu, Z.; Li, J. Peptide-based therapeutic cancer vaccine: Current trends in clinical application. Cell Prolif. 2021, 54, e13025. [Google Scholar] [CrossRef]
- Yang, B.; Jeang, J.; Yang, A.; Wu, T.C.; Hung, C.-F. DNA vaccine for cancer immunotherapy. Hum. Vaccines Immunother. 2014, 10, 3153–3164. [Google Scholar] [CrossRef]
- Mitchell, A.D.; Nair, S.K. RNA transfected dendritic cells as cancer vaccines. Curr. Opin. Mol. Ther. 2000, 2, 76–181. [Google Scholar]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.-J.; Pulendran, B.; Palucka, K. Immunobiology of Dendritic Cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Reardon, D.A.; Mitchell, D.A. The development of dendritic cell vaccine-based immunotherapies for glioblastoma. Semin. Immunopathol. 2017, 39, 225–239. [Google Scholar] [CrossRef]
- Schlesinger, M.J. Heat shock proteins. J. Biol. Chem. 1990, 265, 12111–12114. [Google Scholar] [CrossRef]
- Stetler, R.A.; Gan, Y.; Zhang, W.; Liou, A.K.; Gao, Y.; Cao, G.; Chen, J. Heat shock proteins: Cellular and molecular mechanisms in the central nervous system. Prog. Neurobiol. 2010, 92, 184–211. [Google Scholar] [CrossRef]
- Blachere, N.E.; Li, Z.; Chandawarkar, R.Y.; Suto, R.; Jaikaria, N.S.; Basu, S.; Udono, H.; Srivastava, P.K. Heat Shock Protein–Peptide Complexes, Reconstituted In Vitro, Elicit Peptide-specific Cytotoxic T Lymphocyte Response and Tumor Immunity. J. Exp. Med. 1997, 186, 1315–1322. [Google Scholar] [CrossRef]
- Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Preusser, M.; Stockhammer, G.; Nowosielski, M.; Iglseder, S.; Freyschlag, C.F.; Oberndorfer, S.; et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers 2018, 10, 372. [Google Scholar] [CrossRef]
- Cho, D.-Y.; Yang, W.-K.; Lee, H.-C.; Hsu, D.-M.; Lin, H.-L.; Lin, S.-Z.; Chen, C.-C.; Harn, H.-J.; Liu, C.-L.; Lee, W.-Y.; et al. Adjuvant Immunotherapy with Whole-Cell Lysate Dendritic Cells Vaccine for Glioblastoma Multiforme: A Phase II Clinical Trial. World Neurosurg. 2012, 77, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Peng, J.; Liu, P.; Wu, Q. The Efficacy of Dendritic Cell Vaccine for Newly Diagnosed Glioblastoma: A Meta-analysis of Randomized Controlled Studies. Clin. Neuropharmacol. 2021, 44, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Den.dritic Cell Vaccination with Extension of Survival Among Patients with Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2022, 9, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Litterman, A.J.; Dudek, A.Z.; Largaespada, A.D. Alkylating chemotherapy may exert a uniquely deleterious effect upon neo-antigen-targeting anticancer vaccination. Oncoimmunology 2013, 2, e26294. [Google Scholar] [CrossRef]
- Litterman, A.J.; Zellmer, D.M.; Grinnen, K.L.; Hunt, M.A.; Dudek, A.Z.; Salazar, A.M.; Ohlfest, J.R. Profound Impairment of Adaptive Immune Responses by Alkylating Chemotherapy. J. Immunol. 2013, 190, 6259–6268. [Google Scholar] [CrossRef]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef]
- Chakravarti, A.; Noll, E.; Black, P.M.; Finkelstein, D.F.; Finkelstein, D.M.; Dyson, N.J.; Loeffler, J.S. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J. Clin. Oncol. 2002, 20, 1063–1068. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Yamasaki, F.; Hama, S.; Yahara, K.; Yoshioka, H.; Sugiyama, K.; Arita, K.; Kurisu, K. Expression of survivin in astrocytic tumors: Correlation with malignant grade and prognosis. Cancer Interdiscipl. Int. J. Am. Cancer Soc. 2003, 97, 1077–1083. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2023, 41, 1453–1465. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Binder, D.C.; Ladomersky, E.; Lenzen, A.; Zhai, L.; Lauing, K.L.; Otto-Meyer, S.D.; Lukas, R.V.; Wainwright, D.A. Lessons learned from rindopepimut treatment in patients with EGFRvIII-expressing glioblastoma. Transl. Cancer Res. 2018, 7 (Suppl. S4), S510–S513. [Google Scholar] [CrossRef]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012, 135, 1042–1054. [Google Scholar] [CrossRef]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M.; et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro-Oncology 2019, 21, 923–933. [Google Scholar] [CrossRef]
- Rampling, R.; Peoples, S.; Mulholland, P.J.; James, A.; Al-Salihi, O.; Twelves, C.J.; McBain, C.; Jefferies, S.; Jackson, A.; Stewart, W.; et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2016, 22, 4776–4785. [Google Scholar] [CrossRef]
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W.J. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar]
- Mitchell, D.A.; Xie, W.; Schmittling, R.; Learn, C.; Friedman, A.; McLendon, R.E.; Sampson, J.H. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro-Oncology 2008, 10, 10–18. [Google Scholar] [CrossRef]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., II; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Okamoto, H.; Mineta, T.; Tabuchi, K. Expression of the Wilms’ tumor gene product WT1 in glioblastomas and medulloblastomas. Brain Tumor Pathol. 2004, 21, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.; Hansen, A.R.; Harb, W.A.; Curtis, K.K.; Koga-Yamakawa, E.; Origuchi, M.; Li, Z.; Ertik, B.; Shaib, W.L. Multicenter, Open-Label, Phase I Study of DSP-7888 Dosing Emulsion in Patients with Advanced Malignancies. Target. Oncol. 2021, 16, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Idbaih, A.; Vieito, M.; Tabatabai, G.; Stradella, A.; Ghiringhelli, F.; Burger, M.C.; Mildenberger, I.; Gonzalez, M.; Hervieu, A.; et al. Ctim-17. Eo2401 Therapeutic Vaccine for Patients with Recurrent Glioblastoma: Phase 1/2 Rosalie Study (NCT04116658). Neuro-Oncology 2022, 24 (Suppl. S7), vii63. [Google Scholar] [CrossRef]
- Baudin, E.; Jimenez, C.; Fassnacht, M.; Grisanti, S.; Menke, C.W.; Haak, H.; Subbiah, V.; Capdevila, J.; De La Fouchardiere, C.; Granberg, D.; et al. EO2401, A Novel Microbiome-Derived Therapeutic Vaccine for Patients with Recurrent Glioblastoma: ROSALIE Study; American Society of Clinical Oncology: Alexandria, VA, USA, 2022. [Google Scholar]
- Schmitz-Winnenthal, F.H.; Hohmann, N.; Niethammer, A.G.; Friedrich, T.; Lubenau, H.; Springer, M.; Breiner, K.M.; Mikus, G.; Weitz, J.; Ulrich, A.; et al. Anti-angiogenic activity of VXM01, an oral T-cell vaccine against VEGF receptor 2, in patients with advanced pancreatic cancer: A randomized, placebo-controlled, phase 1 trial. Oncoimmunology 2015, 4, e1001217. [Google Scholar] [CrossRef]
- Wick, W.; Wick, A.; Sahm, F.; Riehl, D.; Von Deimling, A.; Bendszus, M.; Kickingereder, P.; Beckhove, P.; Schmitz-Winnenthal, F.H.; Jungk, C.; et al. VXM01 phase I study in patients with progressive glioblastoma: Final results. J. Clin. Oncol. 2018, 36, 2017. [Google Scholar] [CrossRef]
- Shemesh, C.S.; Hsu, J.C.; Hosseini, I.; Shen, B.-Q.; Rotte, A.; Twomey, P.; Girish, S.; Wu, B. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol. Ther. 2020, 29, 555–570. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Bu, L.-L.; Yan, J.; Wang, Z.; Ruan, H.; Chen, Q.; Gunadhi, V.; Bell, R.B.; Gu, Z. Advances in drug delivery for post-surgical cancer treatment. Biomaterials 2019, 219, 119182. [Google Scholar] [CrossRef]
- Cha, G.D.; Kang, T.; Baik, S.; Kim, D.; Choi, S.H.; Hyeon, T.; Kim, D.-H. Advances in drug delivery technology for the treatment of glioblastoma multiforme. J. Control. Release 2020, 328, 350–367. [Google Scholar] [CrossRef]
- Woodring, R.N.; Gurysh, E.G.; Bachelder, E.M.; Ainslie, K.M. Drug Delivery Systems for Localized Cancer Combination Therapy. ACS Appl. Bio Mater. 2023, 6, 934–950. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Bowen, W.S.; Svrivastava, A.K.; Batra, L.; Barsoumian, H.; Shirwan, H. Current challenges for cancer vaccine adjuvant development. Expert Rev. Vaccines 2018, 17, 207–215. [Google Scholar] [CrossRef]
- Dubensky, T.W., Jr.; Reed, S.G. Adjuvants for cancer vaccines. In Seminars in Immunology; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56. [Google Scholar] [CrossRef]
- Cenciarini, M.; Valentino, M.; Belia, S.; Sforna, L.; Rosa, P.; Ronchetti, S.; D’Adamo, M.C.; Pessia, M. Dexamethasone in Glioblastoma Multiforme Therapy: Mechanisms and Controversies. Front. Mol. Neurosci. 2019, 12, 65. [Google Scholar] [CrossRef]
- Pitter, K.L.; Tamagno, I.; Alikhanyan, K.; Hosni-Ahmed, A.; Pattwell, S.S.; Donnola, S.; Dai, C.; Ozawa, T.; Chang, M.; Chan, T.A.; et al. Corticosteroids compromise survival in glioblastoma. Brain 2016, 139, 1458–1471. [Google Scholar] [CrossRef]
- Swildens, K.X.; Smitt, P.A.E.S.; Bent, M.J.V.D.; French, P.J.; Geurts, M. The effect of dexamethasone on the microenvironment and efficacy of checkpoint inhibitors in glioblastoma: A systematic review. Neuro-Oncology Adv. 2022, 4, vdac087. [Google Scholar] [CrossRef]
- Giles, A.J.; Hutchinson, M.-K.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D.; et al. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 51. [Google Scholar] [CrossRef]
- Iorgulescu, J.B.; Gokhale, P.C.; Speranza, M.C.; Eschle, B.K.; Poitras, M.J.; Wilkens, M.K.; Soroko, K.M.; Chhoeu, C.; and Knott, A.; Gao, Y.; et al. Concurrent Dexamethasone Limits the Clinical Benefit of Immune Checkpoint Blockade in GlioblastomaDexamethasone Limits Anti–PD-1 Benefit for GBM. Clin. Cancer Res. 2021, 27, 276–287. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Bevacizumab (Avastin®) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- Ohm, J.E.; Carbone, D.P. VEGF as a Mediator of Tumor-Associated Immunodeficiency. Immunol. Res. 2001, 23, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Aucouturier, J.; Dupuis, L.; Deville, S.; Ascarateil, S.; Ganne, V. Montanide ISA 720 and 51: A new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev. Vaccines 2002, 1, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Wishart, F.O.; Jackson, L.K. Tetanus Toxoid—The Recall Dose. Canad. J. Public Health/Rev. Can. Sante’e Publique 1948, 39, 181–186. [Google Scholar]
- Alson, D.; Schuyler, S.C.; Yan, B.-X.; Samimuthu, K.; Qiu, J.T. Combination Vaccination with Tetanus Toxoid and Enhanced Tumor-Cell Based Vaccine Against Cervical Cancer in a Mouse Model. Front. Immunol. 2020, 11, 927. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interf. Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Rosenberg, S.A. The Development of New Immunotherapies for the Treatment of Cancer Using Interleukin-2. Ann. Surg. 1988, 208, 121–135. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. Interleukin-2 therapy of cancer-clinical perspectives. Int. Immunopharmacol. 2021, 98, 107836. [Google Scholar] [CrossRef]
- Gillessen, S.; Naumov, Y.N.; Nieuwenhuis, E.E.S.; Exley, M.A.; Lee, F.S.; Mach, N.; Luster, A.D.; Blumberg, R.S.; Taniguchi, M.; Balk, S.P.; et al. CD1d-restricted T cells regulate dendritic cell function and antitumor immunity in a granulocyte–macrophage colony-stimulating factor-dependent fashion. Proc. Natl. Acad. Sci. USA 2003, 100, 8874–8879. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. [Google Scholar] [CrossRef]
- Tang, K.; Wu, Y.-H.; Song, Y.; Bin Yu, B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 68. [Google Scholar] [CrossRef]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2019, 25, 131–147. [Google Scholar] [CrossRef]
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Győrffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660. [Google Scholar] [CrossRef]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; A Wainwright, D. IDO1 in cancer: A Gemini of immune checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Ladomersky, E.; Zhai, L.; Lenzen, A.; Lauing, K.L.; Qian, J.; Scholtens, D.M.; Gritsina, G.; Sun, X.; Liu, Y.; Yu, F.; et al. IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival Against Advanced Glioblastoma. Clin. Cancer Res. 2018, 24, 2559–2573. [Google Scholar] [CrossRef] [PubMed]
- Türeci, Ö.; Löwer, M.; Schrörs, B.; Lang, M.; Tadmor, A.; Sahin, U. Challenges towards the realization of individualized cancer vaccines. Nat. Biomed. Eng. 2018, 2, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Türeci, Ö.; Vormehr, M.; Diken, M.; Kreiter, S.; Huber, C.; Sahin, U. Targeting the Heterogeneity of Cancer with Individualized Neoepitope Vaccines. Clin. Cancer Res. 2016, 22, 1885–1896. [Google Scholar] [CrossRef]
- Bastin, D.J.; Quizi, J.; Kennedy, M.A.; Kekre, N.; Auer, R.C. Current challenges in the manufacture of clinical-grade autologous whole cell vaccines for hematological malignancies. Cytotherapy 2022, 24, 979–989. [Google Scholar] [CrossRef]
- Parney, I.F.; Gustafson, M.; Solseth, M.; Bulur, P.; Peterson, E.T.; Smadbeck, J.B.; Johnson, S.H.; Murphy, S.J.; Vasmatzis, G.; Dietz, A.B. Novel strategy for manufacturing autologous dendritic cell/allogeneic tumor lysate vaccines for glioblastoma. Neuro-Oncol. Adv. 2020, 2, vdaa105. [Google Scholar] [CrossRef]
- Parney, I.F.; Anderson, S.K.; Gustafson, M.P.; Steinmetz, S.; Peterson, E.T.; Kroneman, T.N.; Raghunathan, A.; O’Neill, B.P.; Buckner, J.C.; Solseth, M.; et al. Phase I trial of adjuvant mature autologous dendritic cell/allogeneic tumor lysate vaccines in combination with temozolomide in newly diagnosed glioblastoma. Neuro-Oncol. Adv. 2022, 4, vdac089. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ID | Phase | Design | Patients Enrolled | Diagnosis | Vaccine | Delivery | Adjuvant | Control | Primary Endpoint | Age | Status | Start | Completion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NCT03149003 | Phase 3 | Rand | 236 | rGBM | DSP-7888 | ID | Bevacizumab | Bevacizumab + SOC | DLT, OS | 18+ | Active, not recruiting | 8 December 2017 | 1 November 2023 |
| NCT05100641 | Phase 3 | Rand | 726 | nGBM | AV-GBM-1 | Not listed | GM-CSF | Autologous monocytes | OS | 18+ | Not yet recruiting | 1 March 2022 | 1 March 2027 |
| NCT04277221 | Phase 3 | Rand | 118 | rGBM | ADCTA | SC | Bevacizumab | Bevacizumab + SOC | OS | 18–70 | Unknown status | 19 September 2019 | 31 December 2022 |
| NCT02455557 | Phase 2 | Single Arm | 66 | nGBM | SurVaxM | SC | Montanide ISA 51 and Sargramostim | - | PFS6 | 18+ | Active, not recruiting | 4 May 2015 | 30 December 2023 |
| NCT01204684 | Phase 2 | Rand | 60 | n/rGBM | ATL-DC | ID | Resiquimod or Poly-ICLC | - | Most effective combination | 18–70 | Active, not recruiting | 8 October 2010 | 31 January 2025 |
| NCT03400917 | Phase 2 | Single Arm | 55 | nGBM | AV-GBM-1 | Not listed | GM-CSF | - | OS | 18–70 | Active, not recruiting | 20 June 2018 | 1 February 2023 |
| NCT04523688 | Phase 2 | Single Arm | 28 | GBM | ATL-DC | ID | - | - | PFS, AEs | 18+ | Not yet recruiting | 1 March 2021 | 1 December 2025 |
| NCT04888611 | Phase 2 | Rand | 40 | rGBM | GSC-DCV | Not listed | Camrelizumab | Camrelizumab alone | OS, PFS | 18–70 | Recruiting | 26 October 2021 | 1 May 2024 |
| NCT02465268 | Phase 2 | Rand | 175 | nGBM | CMV-DC | SC | GM-CSF | Autologous monocytes | OS | 18+ | Recruiting | 1 August 2016 | 1 June 2024 |
| NCT04280848 | Phase 2 | Non-Rand | 56 | nGBM | UCPVax | SC | - | - | Immunogenicity | 18–75 | Recruiting | 26 May 2020 | 1 May 2023 |
| NCT03395587 | Phase 2 | Rand | 136 | nGBM | ATL-DC | ID | - | SOC | OS | 18+ | Recruiting | 6 March 2018 | 6 June 2025 |
| NCT05163080 | Phase 2 | Rand | 265 | nGBM | SurVaxM | SC | Montanide and Sargramostim | Montanide and Sargramostim | OS | 18+ | Recruiting | 18 November 2021 | 18 April 2024 |
| NCT03382977 | Phase 1/2 | Non-Rand | 98 | rGBM | VBI-1901 | ID | GM-CSF | SOC | DLT, AEs | 18+ | Active, not recruiting | 6 December 2017 | 1 August 2025 |
| NCT03665545 | Phase 1/2 | Rand | 18 | rGBM | IMA950 | SC | Poly-ICLC +/− Pembrolizumab | - | AEs | 18+ | Active, not recruiting | 25 October 2018 | 31 December 2023 |
| NCT03750071 | Phase 1/2 | Single Arm | 30 | rGBM | VXM01 | Not listed | Avelumab | - | AEs | 18+ | Active, not recruiting | 21 November 2018 | 31 December 2022 |
| NCT04116658 | Phase 1/2 | Non-Rand | 52 | rGBM | EO2401 | Not listed | Nivolumab +/− Bevacizumab | - | AEs | 18+ | Recruiting | 13 July 2020 | 1 August 2023 |
| NCT02649582 | Phase 1/2 | Single Arm | 20 | nGBM | auto-WT1-DC | ID | - | - | OS | 18+ | Recruiting | 1 December 2015 | 1 December 2024 |
| NCT04801147 | Phase 1/2 | Single Arm | 76 | nGBM | ATL-DC | ID | - | - | PFS12 | 18–70 | Recruiting | 1 June 2010 | 1 December 2023 |
| NCT04388033 | Phase 1/2 | Single Arm | 10 | nGBM | ATL-DC | ID | IL-12 | - | AEs, PFS6 | 18–75 | Recruiting | 1 December 2020 | 1 December 2023 |
| NCT04015700 | Phase 1 | Single Arm | 9 | nGBM | GNOS-PV01 | Not listed | INO-9012 (IL-12) | - | DLT, Feasibility | 18+ | Active, not recruiting | 14 July 2020 | 13 April 2023 |
| NCT03223103 | Phase 1 | Single Arm | 13 | nGBM | MTA-based Personalized Vaccine | Not listed | Poly-ICLC | - | DLT | 18+ | Active, not recruiting | 1 March 2018 | 1 May 2023 |
| NCT04642937 | Phase 1 | Sequential | 24 | rGBM | GBM6-AD (Allogeneic TL) | Not listed | hP1A8 + Imiquimod | - | MTD | 18+ | Active, not recruiting | 1 December 2020 | 1 November 2023 |
| NCT00639639 | Phase 1 | Single Arm | 42 | nGBM | CMV-DC (auto) | ID | tetanus toxoid | - | Feasibility | 18+ | Active, not recruiting | 1 January 2006 | 1 December 2022 |
| NCT04741984 | Phase 1 | Sequential | 27 | nGBM | MT-201-GBM | IV | - | - | MTD, Immunogenicity | 18+ | Not yet recruiting | 1 October 2022 | 1 August 2025 |
| NCT05283109 | Phase 1 | Single Arm | 36 | nGBM | P30-EPS (P30-linked EphA2, CMV pp65, survivin) | Not listed | Hiltonol | - | DLT | 18+ | Not yet recruiting | 1 November 2022 | 1 February 2028 |
| NCT04968366 | Phase 1 | Single Arm | 10 | nGBM | ATL-DC (with multiple tumor neoantigen peptides) | ID | - | - | AEs | 18–75 | Recruiting | 30 July 2021 | 1 August 2024 |
| NCT04573140 | Phase 1 | Single Arm | 28 | nGBM | RNA-LP | IV | - | - | Feasibility, DLT, MTD | 21+ | Recruiting | 26 October 2021 | 1 July 2027 |
| NCT04963413 | Phase 1 | Single Arm | 10 | nGBM | CMV-DC (auto) | Not listed | GM-CSF | - | Feasibility | 18–90 | Recruiting | 13 January 2022 | 1 May 2025 |
| NCT02287428 | Phase 1 | Rand | 56 | nGBM | NeoVax | Not listed | Pembrolizumab | - | AEs, Feasibility | 18+ | Recruiting | 1 November 2014 | 1 January 2026 |
| NCT04201873 | Phase 1 | Rand | 40 | rGBM | ATL-DC | ID | Pembrolizumab, Poly-ICLC | IV Placebo | Immunogenicity, AEs | 18+ | Recruiting | 8 January 2020 | 1 August 2025 |
| NCT04552886 | Phase 1 | Non-Rand | 24 | nGBM | ATL-DC (Th-1 specific) | Not listed | - | - | AEs | 18+ | Recruiting | 11 October 2021 | 31 December 2025 |
| NCT04842513 | Phase 1 | Single Arm | 15 | nGBM | Multipeptide vaccine | SC | XS15 | - | AEs, Immunogenicity | 18+ | Recruiting | 3 May 2021 | 2 May 2024 |
| NCT05557240 | Phase 1 | Single Arm | 10 | nGBM | NeoPep Vaccine1/2 (NPVAC1/2) | Not listed | Poly-ICLC | - | AEs | 18–70 | Recruiting | 13 September 2022 | 12 August 2025 |
| NCT03360708 | Early Phase 1 | Single Arm | 20 | rGBM | ATL-DC + Allo GBM lysate | ID | - | - | AEs, Feasibility | 18+ | Active, not recruiting | 3 June 2019 | 1 June 2023 |
| NCT01957956 | Early Phase 1 | Single Arm | 21 | nGBM | auto-DC + Allo GBM lysate | ID | - | - | AEs | 18+ | Active, not recruiting | 11 November 2013 | 15 November 2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neth, B.J.; Webb, M.J.; Parney, I.F.; Sener, U.T. The Current Status, Challenges, and Future Potential of Therapeutic Vaccination in Glioblastoma. Pharmaceutics 2023, 15, 1134. https://doi.org/10.3390/pharmaceutics15041134
Neth BJ, Webb MJ, Parney IF, Sener UT. The Current Status, Challenges, and Future Potential of Therapeutic Vaccination in Glioblastoma. Pharmaceutics. 2023; 15(4):1134. https://doi.org/10.3390/pharmaceutics15041134
Chicago/Turabian StyleNeth, Bryan J., Mason J. Webb, Ian F. Parney, and Ugur T. Sener. 2023. "The Current Status, Challenges, and Future Potential of Therapeutic Vaccination in Glioblastoma" Pharmaceutics 15, no. 4: 1134. https://doi.org/10.3390/pharmaceutics15041134
APA StyleNeth, B. J., Webb, M. J., Parney, I. F., & Sener, U. T. (2023). The Current Status, Challenges, and Future Potential of Therapeutic Vaccination in Glioblastoma. Pharmaceutics, 15(4), 1134. https://doi.org/10.3390/pharmaceutics15041134