
“Click-to-Clear”: A Strategy to Minimize Radioactivity from the Blood Pool Utilizing Staudinger Ligation

 ,
, 
 ,
, 
Abstract
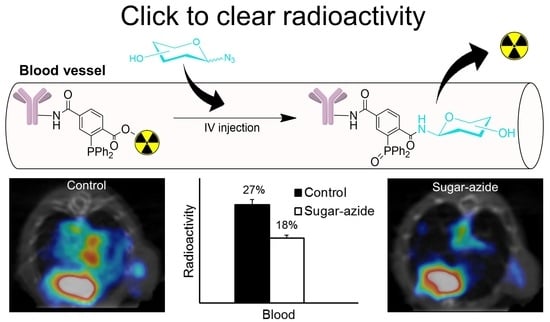
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. General Information
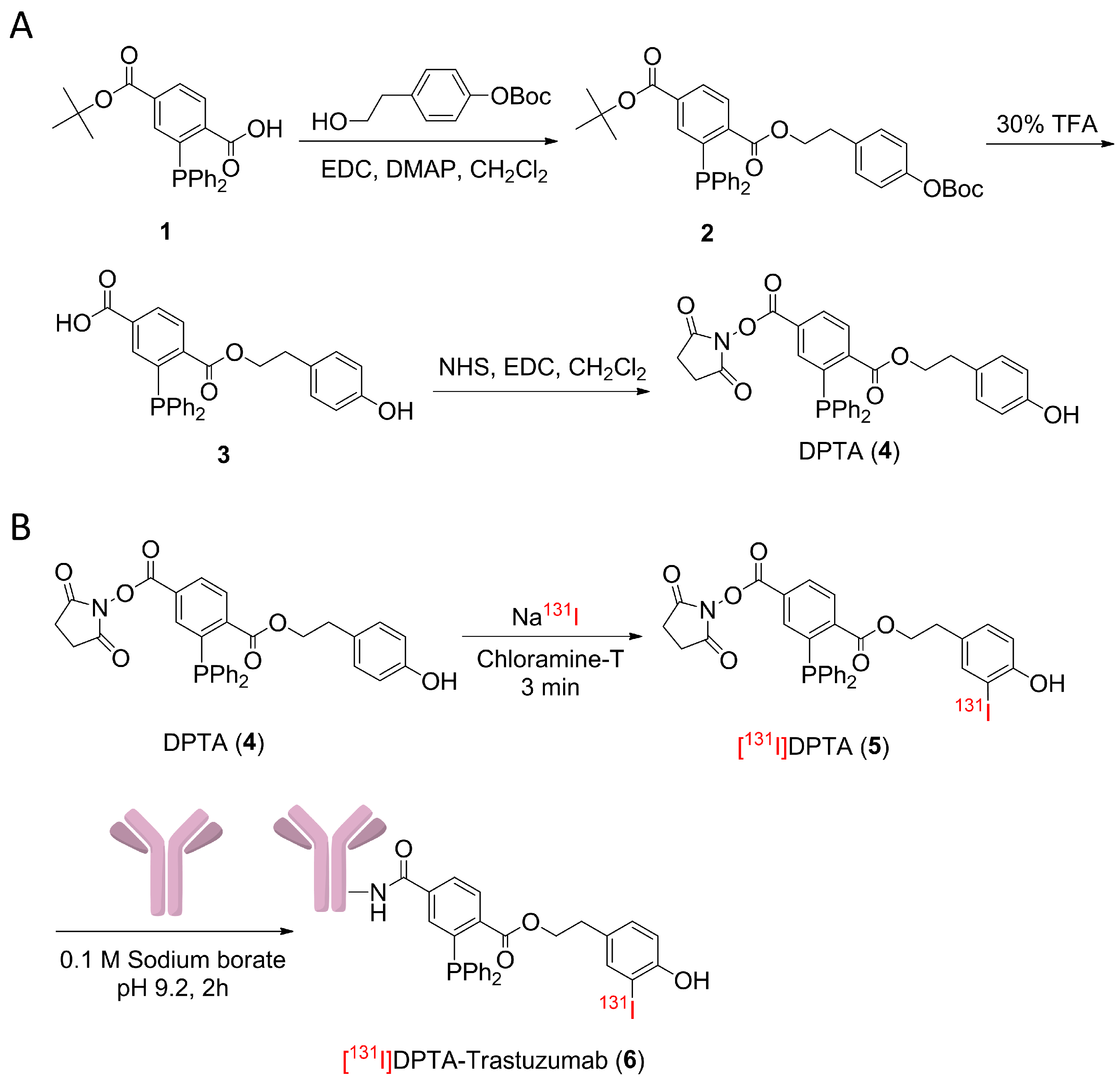
2.2. Synthesis and Characterization
2.2.1. 1-(4-((Tert-butoxycarbonyl)oxy)phenethyl) 4-(tert-butyl) 2-(diphenylphosphaneyl) terephthalate (Compound 2)
2.2.2. 3-(Diphenylphosphaneyl)-4-((4-hydroxyphenethoxy)carbonyl)benzoic Acid (Compound 3)
2.2.3. 4-(2,5-Dioxopyrrolidin-1-yl) 1-(4-hydroxyphenethyl) 2-(diphenylphosphaneyl)terephthalate (DPTA) (Compound 4)
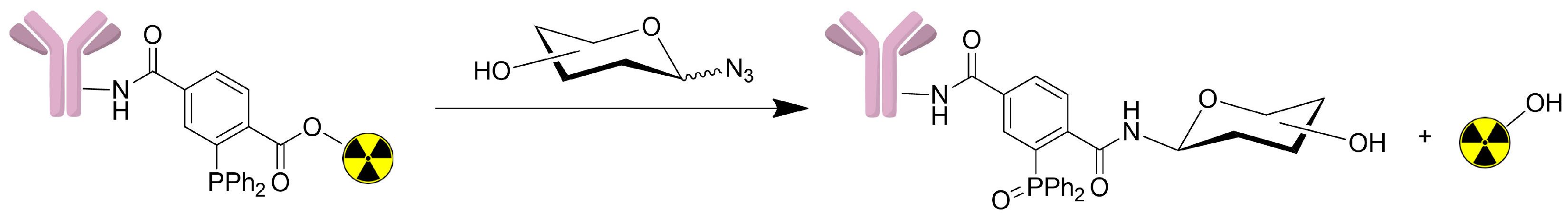
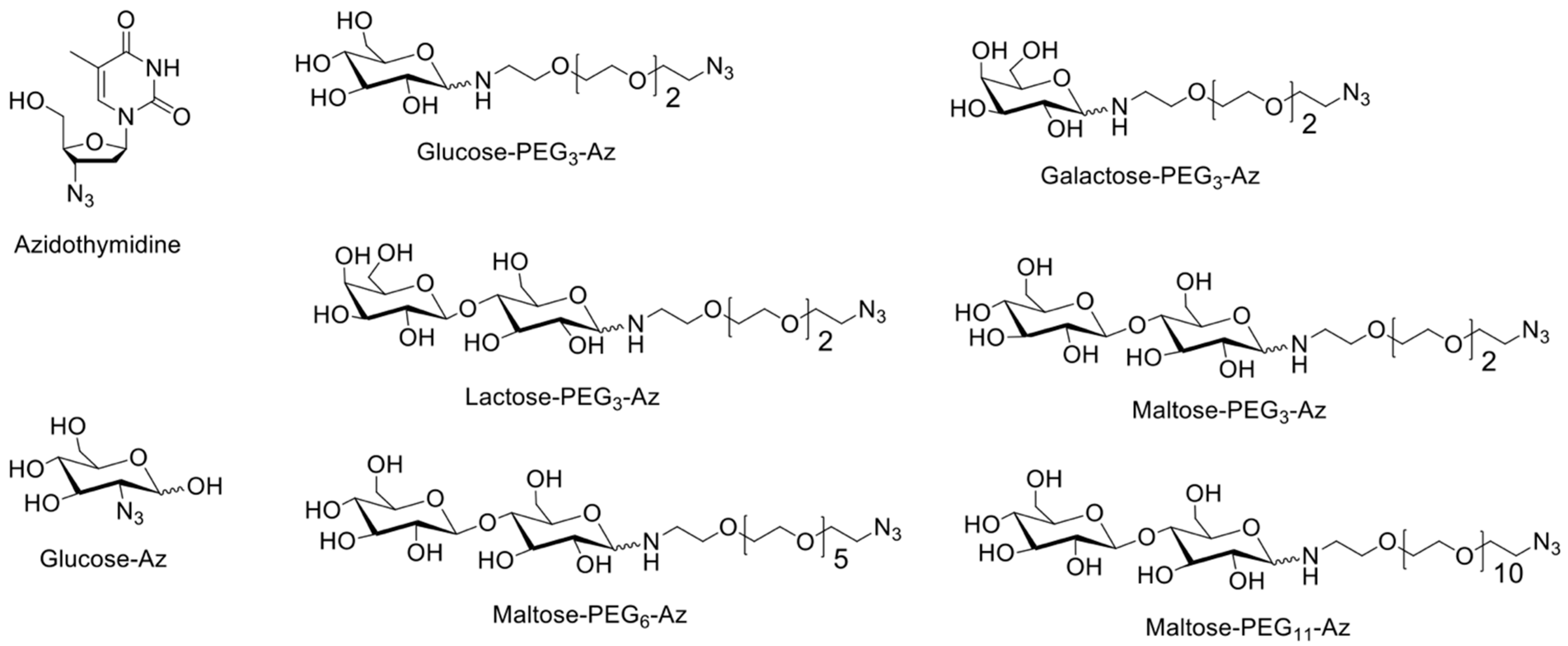
2.2.4. Synthesis of Sugar Azides
- Glucose-PEG3-azide: D-(+)-glucose (180 mg, 1.0 mmol); 11-azido-3,6,9-trioxaundecan-1-amine (240 mg, 1.1 mmol, 1.1 equivalent); glucose-PEG3-azide final yield (300 mg, 79%). 1H NMR (D2O, 500 MHz): δ 2.78–2.83 (m), 2.92 (t, J = 5 Hz), 2.98–3.02 (m), 3.11–3.17 (m), 3.28–3.31 (m), 3.38–3.43 (m), 3.57–3.82 (m), 3.93 (d, J = 9 Hz), 4.55 (d, J = 8 Hz), and 5.14 (d, J = 3.5) ppm; 13C NMR (D2O, 125 MHz): δ 39.51, 50.17, 60.78, 60.94, 69.21, 69.23, 69.40, 69.55, 69.59, 69.62, 69.65, 69.91, 70.16, 71.44, 71.50, 72.77, 72.96, 74.16, 75.78, 75.95, 76.75, 76.79, 92.10, and 95.93 ppm; HRMS (FAB): m/z calculated for C14H28N4O8 [M+H]+: 381.1985; found: 381.2041.
- Galactose-PEG3-azide: D-(+)-galactose (500 mg, 1.0 mmol); 11-azido-3,6,9-trioxaundecan-1-amine (666 mg, 1.1 mmol, 1.1 equivalent); galactose-PEG3-azide final yield (308 mg, 29%). 1H NMR (D2O, 500 MHz): δ 3.09 (t, J = 5 Hz), 2.98–3.02 (m), 3.38–3.44 (m), 3.54–3.57 (m), 3.61(d, J = 4Hz), 3.64–3.68 (m), 3.74 (d, J = 15 Hz), 3.83 (d, J = 5 Hz), 3.84 (d, J = 5 Hz), 3.9–4.01 (m), 4.49 (d, J = 8 Hz), and 5.17 (d, J = 3.5 Hz) ppm; 13C NMR (D2O, 125 MHz): δ 39.20, 50.14, 60.97, 61.17, 68.34, 68.74, 69.16, 69.21, 69.30, 69.45, 69.49, 69.54, 69.56, 69.63, 70.46, 71.87, 72.79, 75.14, 92.27, and 96.44 ppm; HRMS (FAB): m/z calculated for C14H28N4O8 [M+H]+: 381.1985; found: 381.1983.
- Lactose-PEG3-azide: α-lactose (1 g, 1.0 mmol); 11-azido-3,6,9-trioxaundecan-1-amine (0.7 g, 1.1 mmol, 1.1 equivalent.); lactose-PEG3-azide final yield (1.1 g, 70%). 1H NMR (D2O, 400 MHz): δ 2.73–2.79 (m), 2.92–2.98 (m), 3.10–3.15 (m), 3.37–3.49 (m), 3.51–3.84 (m), 3.92 (d, J = 8 Hz), 4.32 (d, J = 8 Hz), 4.53 (d, J = 8 Hz), and 5.09 (d, J = 3.6 Hz) ppm; 13C NMR (D2O, 100 MHz): δ 39.90, 44.55, 50.49, 61.39, 68.90, 69.58, 69.73, 69.76, 69.86, 69.88, 69.93, 70.00, 70.45, 70.48, 71.31, 72.87, 72.97, 74.17, 74.72, 75.14, 75.70, 75.93, 78.64, 78.98, 89.87, 92.17, 96.12, and 103.26 ppm; HRMS (FAB): m/z calculated for C20H38N4O13 [M+H]+: 543.2514; found: 543.2514.
- Maltose-PEG3-azide: D-(+)-maltose (350 mg, 1.0 mmol); 11-azido-3,6,9-trioxaundecan-1-amine (245.5 mg, 1.1 mmol, 1.1 equivalent); maltose-PEG3-azide final yield (450 mg, 81%). 1H NMR (D2O, 400 MHz): δ 2.74–2.77 (m), 2.91–2.97 (m), 3.08–3.15 (m), 3.28 (t, J = 9.2 Hz), 3.36–3.76 (m), 3.91 (d, J = 8 Hz), 4.51 (d, J = 8 Hz), 5.08–5.09 (m), and 5.27 (s) ppm; 13C NMR (D2O, 100 MHz): δ 39.84, 44.52, 50.48, 60.81, 69.58, 69.73, 69.88, 69.93, 70.00, 70.28, 70.48, 71.63, 72.00, 72.09, 73.01, 73.18, 73.21, 73.58, 74.34, 74.89, 75.67, 76.56, 77.00, 77.22, 77.40, 77.56, 89.86, 92.23, 93.12, and 99.90 ppm; HRMS (FAB): m/z calculated for C20H38N4O13 [M+H]+: 543.2514; found: 543.2518.
- Maltose-PEG6-azide: D-(+)-maltose (100 mg, 1.0 mmol); O-(2-Aminoethyl)-O′-(2-azidoethyl) pentaethylene glycol (112.6 mg, 1.1 mmol, 1.1 equivalent); maltose-PEG6-azide final yield (170 mg, 86%). 1H NMR (D2O, 400 MHz): δ 2.84–2.88 (m), 3.09–3.12 (m), 3.17 (t, J = 7.2 Hz), 3.28–3.45 (m), 3.48 (dd, J = 7.6, 4.8 Hz), 3.53 (t, J = 7.2 Hz), 3.67–3.68 (m), 3.69–3.87 (m), 3.91 (d, J = 7.2 Hz), and 5.18 (d, J = 3.2 Hz) ppm; 13C NMR (D2O, 125 MHz): δ 39.20, 44.19, 50.16, 60.51, 60.60, 60.74, 60.90, 69.21, 69.23, 69.34, 69.36, 69.47, 69.52, 69.55, 69.58, 69.63, 69.96, 70.15, 71.30, 71.68, 71.72, 71.78, 72.67, 72.69, 72.82, 72.86, 72.89, 73.22, 74.01, 74.57, 75.35, 76.30, 76.81, 77.05, 77.20, 77.23, 89.55, 91.89, 95.78, and 99.62 ppm; HRMS (FAB): m/z calculated for C26H50N4O16 [M+H]+: 675.3300; found: 675.3298.
- Maltose-PEG11-azide: D-(+)-maltose (100 mg, 1.0 mmol); O-(2-Aminoethyl)-O′-[2-azidoethyl] decaethylene glycol (140.9 mg, 1.1 mmol, 1.1 equivalent); maltose-PEG11-azide final yield (132 mg, 51%). 1H NMR (D2O, 400 MHz): δ 2.84–2.88 (m), 3.09–3.18 (m), 3.28–3.33 (m), 3.41 (t, J = 4 Hz), 3.48 (dd, J = 7.6, 3.2 Hz), 3.53 (t, J = 7.2 Hz), 3.62–3.69 (m), 3.82 (t, J = 4 Hz), 3.91 (d, J = 7.2 Hz), and 5.18 (d, J = 3.2 Hz) ppm; 13C NMR (D2O, 125 MHz): δ 39.23, 44.23, 50.18, 60.54, 60.61, 60.76, 60.91, 69.38, 69.61, 69.99, 70.14, 71.32, 71.71, 71.75, 71.81, 72.69, 72.83, 72.89, 72.92, 73.23, 74.02, 75.37, 76.22, 76.89, 77.20, 77.31, 89.58, 91.91, 95.81, and 99.67 ppm; HRMS (FAB): m/z calculated for C36H70N4O21 [M+H]+: 895.4611; found: 895.4608.
2.2.5. Synthesis of 4-(2-Hydroxyethyl)-2-iodophenol
2.3. Radiolabeling and Bioconjugation
2.4. In Vitro Stability Studies
2.5. In Vitro Staudinger Ligation
2.6. Effect of Solvent on In Vitro Staudinger Ligation
2.7. Cell Viability Assay
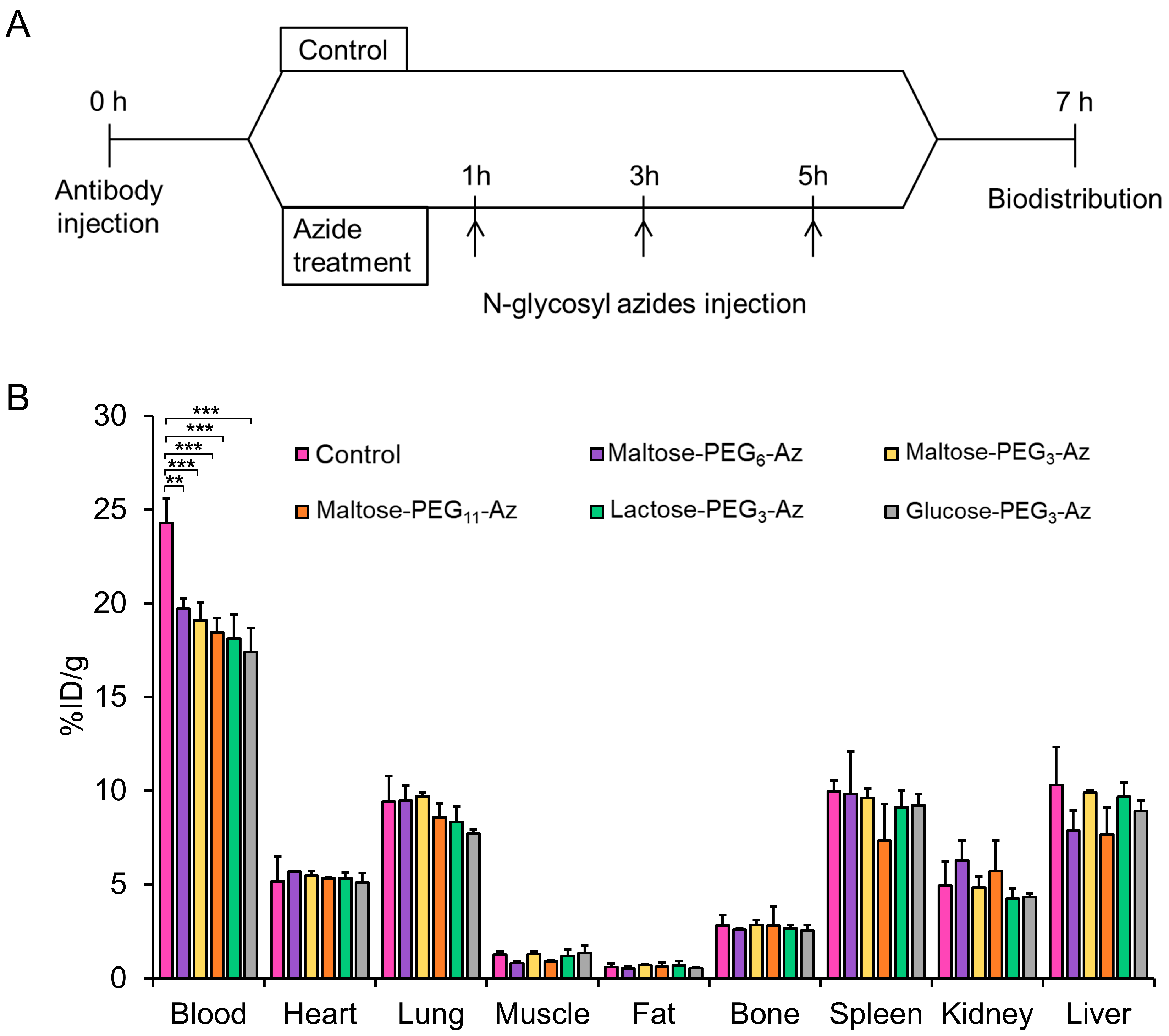
2.8. Animal Studies
2.9. Ex Vivo Analysis of Staudinger Ligation in Blood
2.10. Biodistribution Studies
2.11. SPECT Imaging
2.12. Statistical Analysis
3. Results
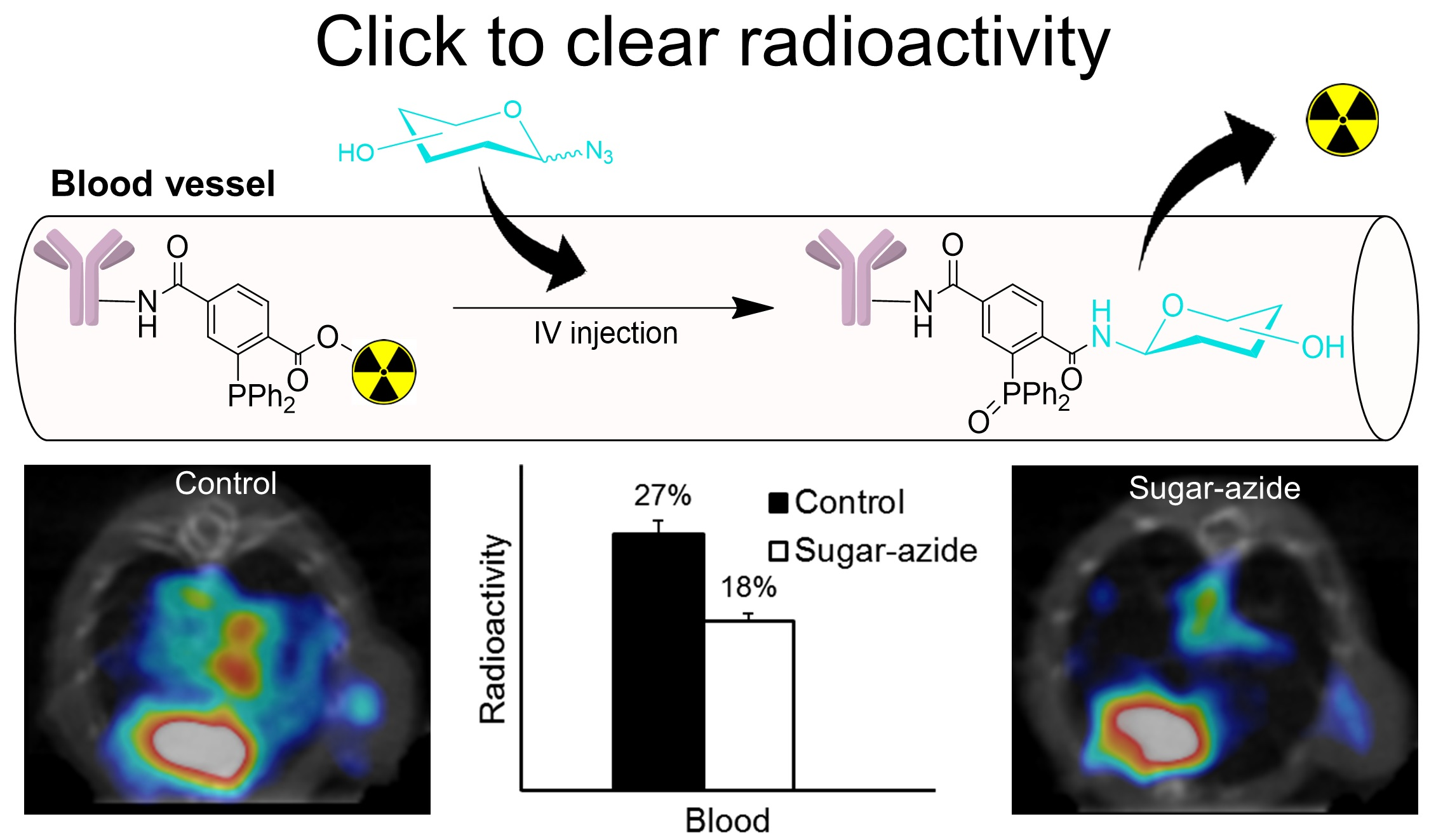
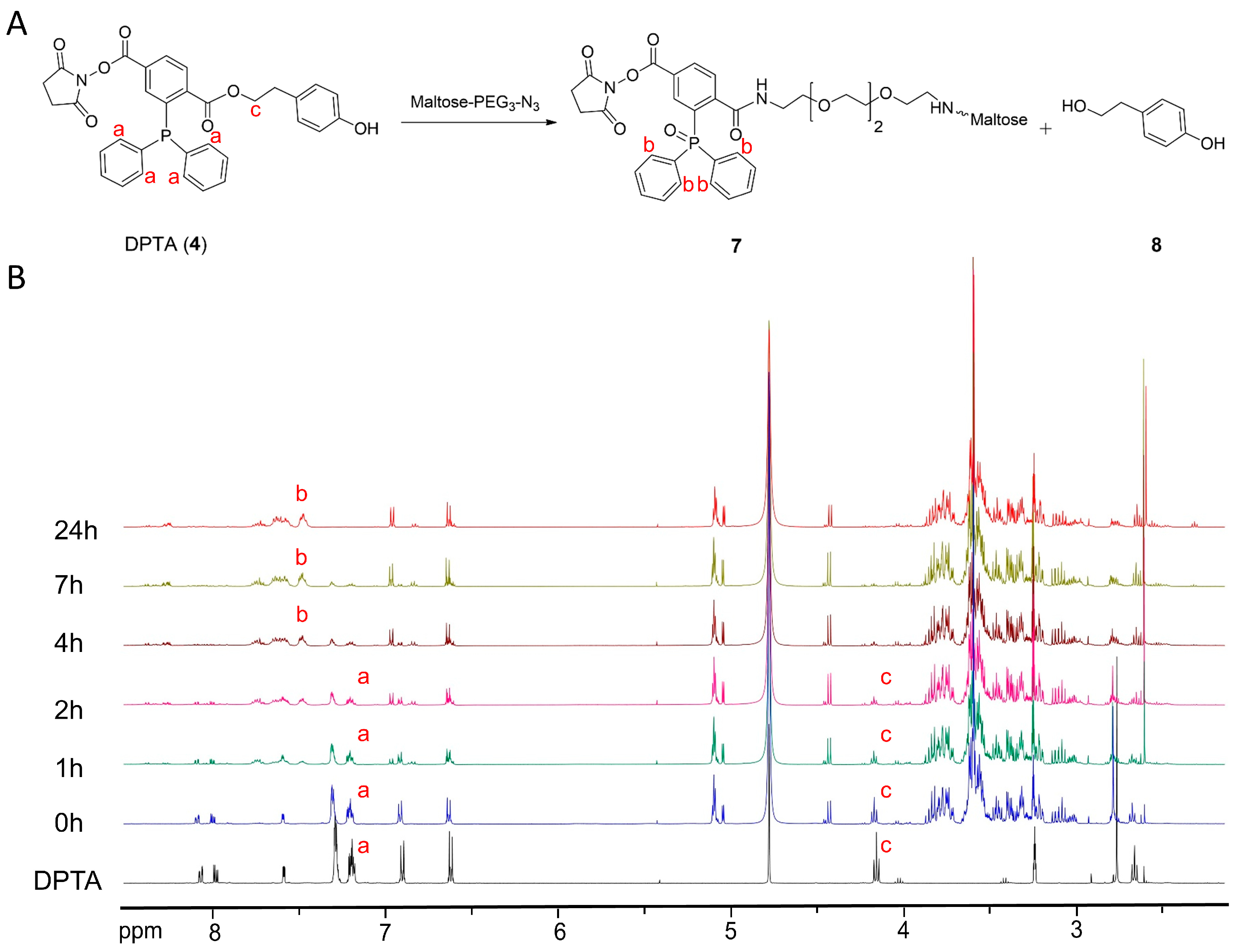
3.1. Synthesis of Prosthetic Group DPTA and Characterization
3.2. Synthesis of Sugar Azides
3.3. 1H-NMR Kinetic Study
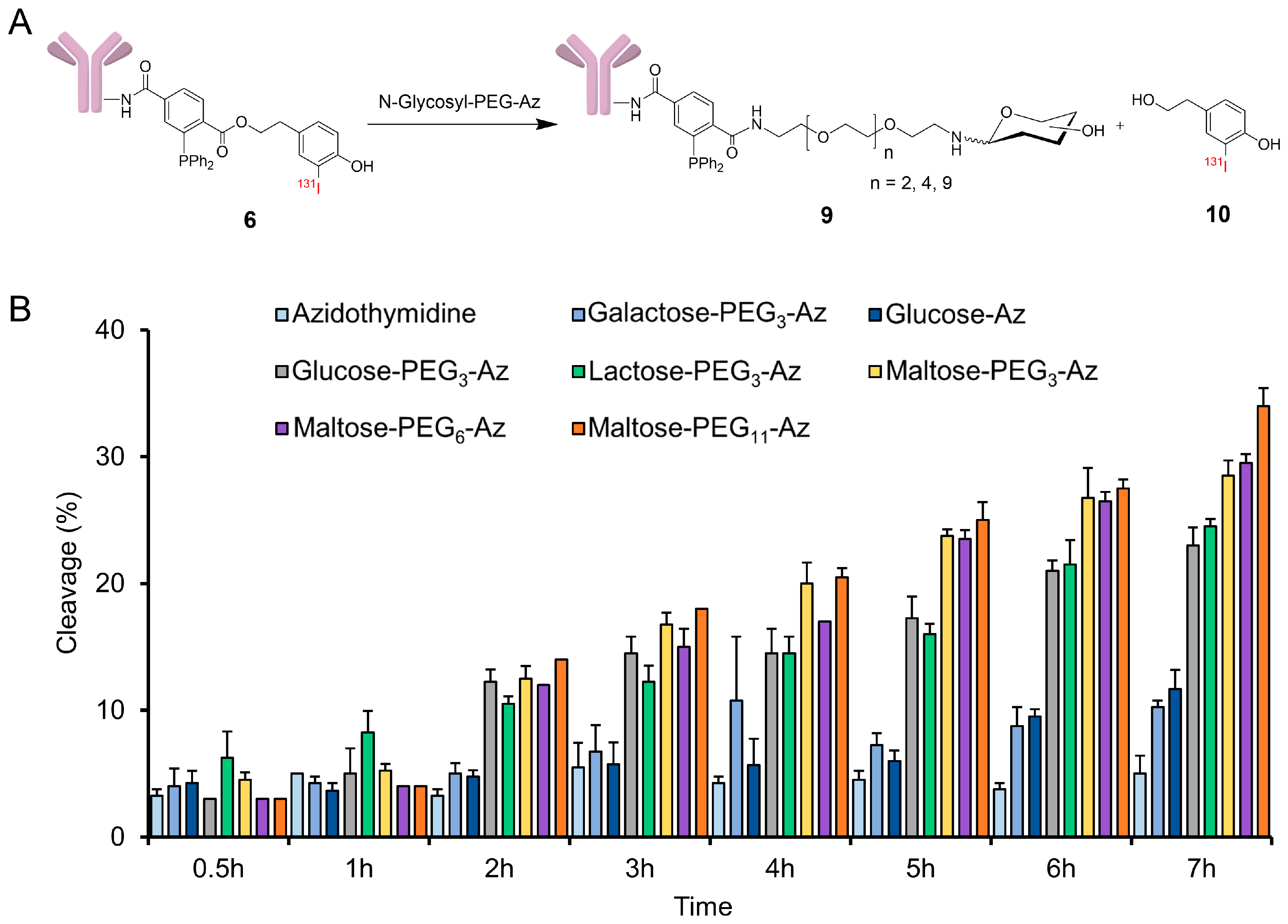
3.4. Radioiodination and Bioconjugation
3.5. Synthesis and Radiolabeling of 4-(2-Hydroxyethyl)-2-iodophenol
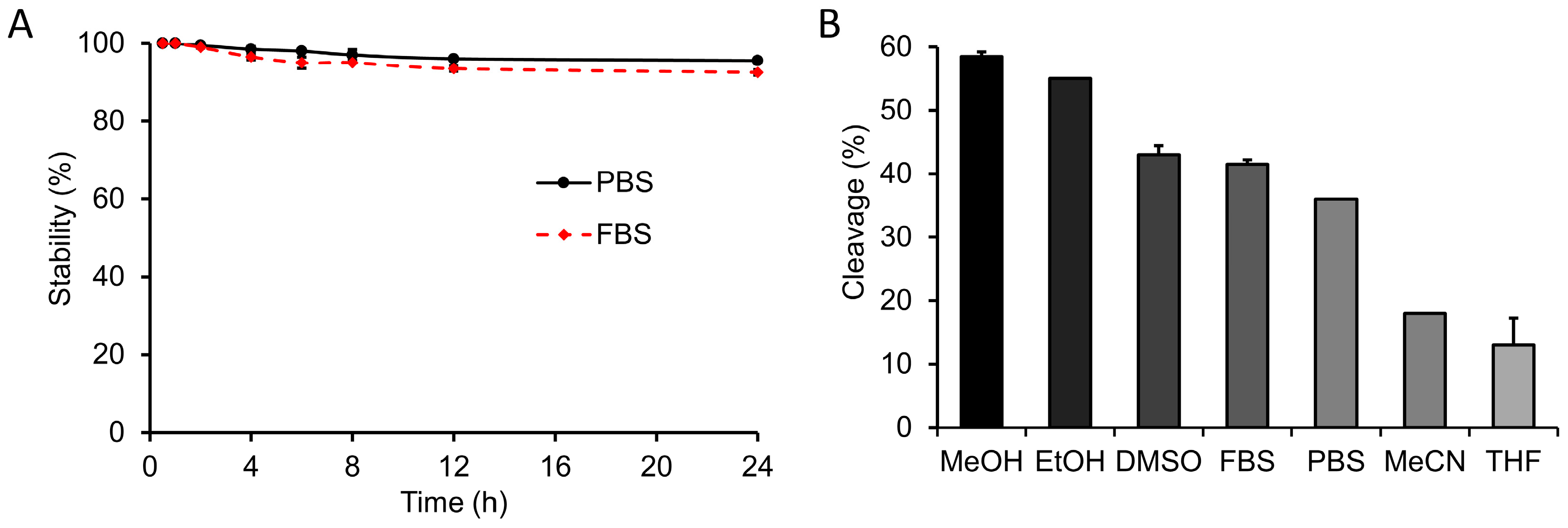
3.6. In Vitro Stability
3.7. Effect of Solvent on In Vitro Staudinger Ligation
3.8. Cleavage of Sugar Azides
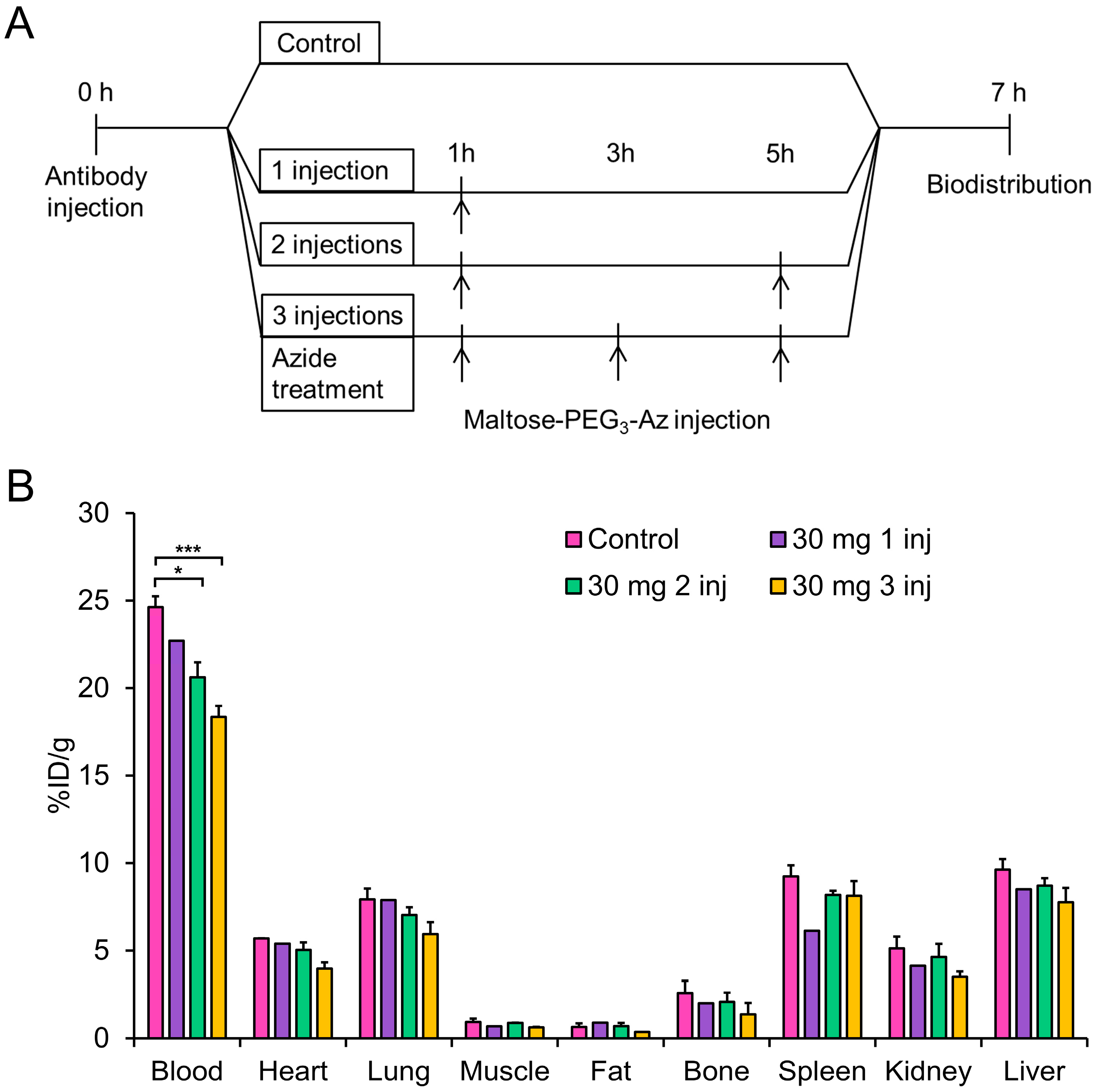
3.9. Blood Analysis of In Vivo Staudinger Ligation
3.10. Biodistribution Studies
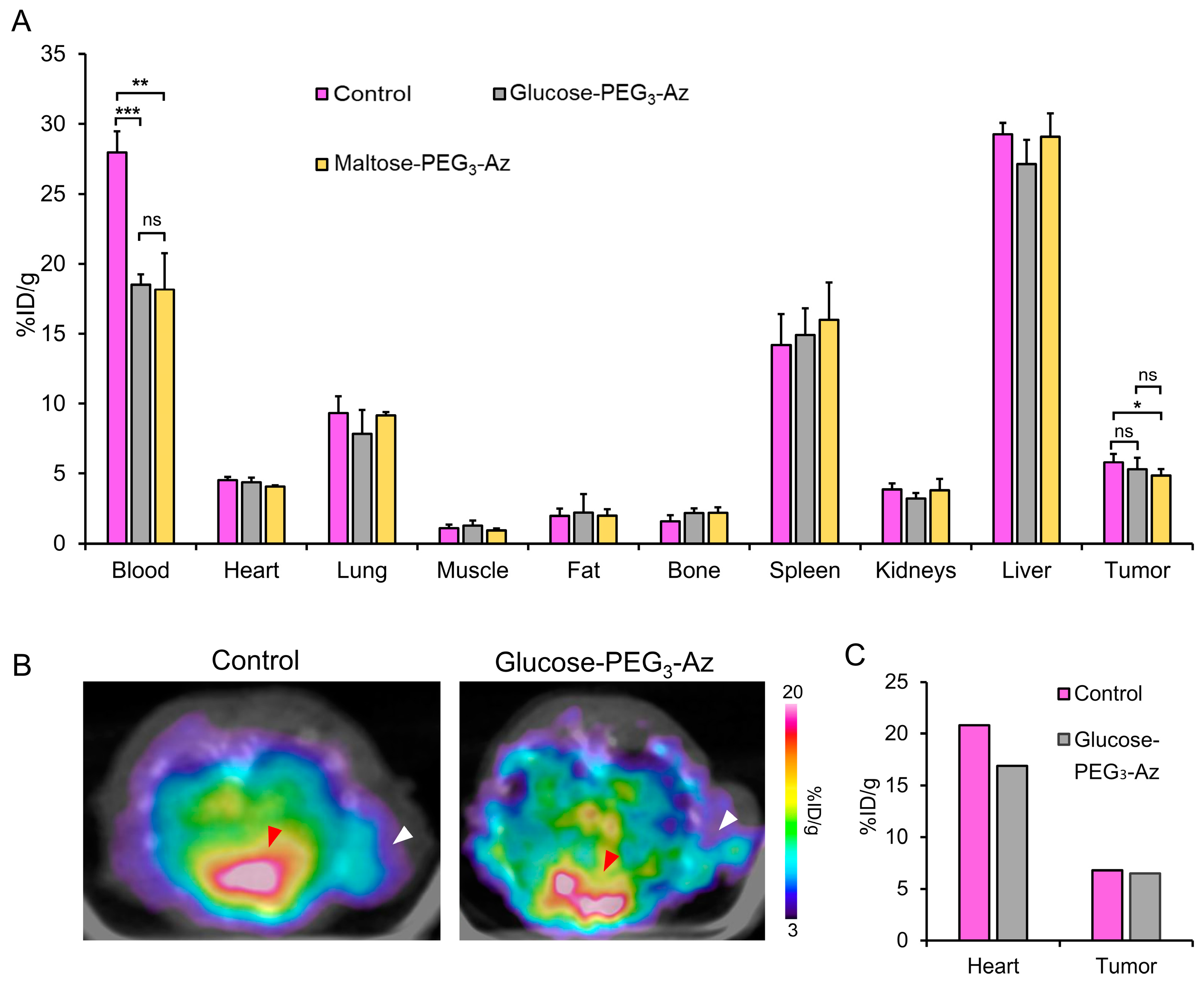
3.11. SPECT Imaging and Biodistribution Studies in Tumor Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bertozzi, C.R. A decade of bioorthogonal chemistry. Acc. Chem. Res. 2011, 44, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.A.; Bertozzi, C.R. Chemistry in living systems. Nat. Chem. Biol. 2005, 1, 13–21. [Google Scholar] [CrossRef]
- Kang, D.; Lee, S.; Kim, J. Bioorthogonal click and release: A general, rapid, chemically revertible bioconjugation strategy employing enamine N-oxides. Chem 2022, 8, 2260–2277. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, S.T.; Bertozzi, C.R. Metabolic labeling of glycans with azido sugars and subsequent glycan-profiling and visualization via Staudinger ligation. Nat. Protoc. 2007, 2, 2930–2944. [Google Scholar] [CrossRef]
- Lang, K.; Chin, J.W. Bioorthogonal reactions for labeling proteins. ACS Chem. Biol. 2014, 9, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Rossin, R.; Renart Verkerk, P.; van den Bosch, S.M.; Vulders, R.; Verel, I.; Lub, J.; Robillard, M.S. In vivo chemistry for pretargeted tumor imaging in live mice. Angew. Chem. Int. Ed. 2010, 49, 3375–3378. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, N.K.; Upadhyay, R.; Haun, J.B.; Hilderbrand, S.A.; Weissleder, R. Fast and sensitive pretargeted labeling of cancer cells through a tetrazine/trans-cyclooctene cycloaddition. Angew. Chem. Int. Ed. 2009, 48, 7013–7016. [Google Scholar] [CrossRef]
- Li, J.; Chen, P.R. Development and application of bond cleavage reactions in bioorthogonal chemistry. Nat. Chem. Biol. 2016, 12, 129–137. [Google Scholar] [CrossRef]
- Azoulay, M.; Tuffin, G.; Sallem, W.; Florent, J.-C. A new drug-release method using the Staudinger ligation. Bioorg. Med. Chem. Lett. 2006, 16, 3147–3149. [Google Scholar] [CrossRef]
- Brakel, R.v.; Vulders, R.C.; Bokdam, R.J.; Grüll, H.; Robillard, M.S. A doxorubicin prodrug activated by the staudinger reaction. Bioconjug. Chem. 2008, 19, 714–718. [Google Scholar] [CrossRef]
- Versteegen, R.M.; Rossin, R.; ten Hoeve, W.; Janssen, H.M.; Robillard, M.S. Click to release: Instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem. Int. Ed. 2013, 52, 14112–14116. [Google Scholar] [CrossRef]
- Jiménez-Moreno, E.; Guo, Z.; Oliveira, B.L.; Albuquerque, I.S.; Kitowski, A.; Guerreiro, A.; Boutureira, O.; Rodrigues, T.; Jiménez-Osés, G.; Bernardes, G.J. Vinyl Ether/Tetrazine Pair for the Traceless Release of Alcohols in Cells. Angew. Chem. 2017, 129, 249–253. [Google Scholar] [CrossRef]
- Luo, W.; Gobbo, P.; Gunawardene, P.N.; Workentin, M.S. A Fluorogenic Gold Nanoparticle (AuNP) Substrate: A Model for the Controlled Release of Molecules from AuNP Nanocarriers via Interfacial Staudinger-Bertozzi Ligation. Langmuir 2017, 33, 1908–1913. [Google Scholar] [CrossRef] [PubMed]
- Saxon, E.; Bertozzi, C.R. Cell surface engineering by a modified Staudinger reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef]
- Saxon, E.; Armstrong, J.I.; Bertozzi, C.R. A “traceless” Staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2000, 2, 2141–2143. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, C.; Wehl, I.; Jung, N.; Schepers, U.; Brase, S. The Staudinger Ligation. Chem. Rev. 2020, 120, 4301–4354. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Prescher, J.A.; Sletten, E.M.; Baskin, J.M.; Miller, I.A.; Agard, N.J.; Lo, A.; Bertozzi, C.R. Copper-free click chemistry in living animals. Proc. Natl. Acad. Sci. USA 2010, 107, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Mamat, C.; Gott, M.; Steinbach, J. Recent progress using the Staudinger ligation for radiolabeling applications. J. Label. Comp. Radiopharm. 2018, 61, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Hansen, R.J.; Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 2004, 93, 2645–2668. [Google Scholar] [CrossRef] [PubMed]
- Kozak, R.W.; Waldmann, T.A.; Atcher, R.W.; Gansow, O.A. Radionuclide-conjugated monoclonal antibodies: A synthesis of immunology, inorganic chemistry and nuclear science. Trends Biotechnol. 1986, 4, 259–264. [Google Scholar] [CrossRef]
- Chomet, M.; van Dongen, G.A.M.S.; Vugts, D.J. State of the Art in Radiolabeling of Antibodies with Common and Uncommon Radiometals for Preclinical and Clinical Immuno-PET. Bioconjug. Chem. 2021, 32, 1315–1330. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.M.; Gotthardt, M.; Becker, W.; Behe, M. Radioiodination of monoclonal antibodies, proteins and peptides for diagnosis and therapy. A review of standardized, reliable and safe procedures for clinical grade levels kBq to GBq in the Gottingen/Marburg experience. Nuklearmedizin 2002, 41, 71–79. [Google Scholar] [PubMed]
- Koppe, M.J.; Postema, E.J.; Aarts, F.; Oyen, W.J.; Bleichrodt, R.P.; Boerman, O.C. Antibody-guided radiation therapy of cancer. Cancer Metastasis Rev. 2005, 24, 539–567. [Google Scholar] [CrossRef] [PubMed]
- Knox, S.J. Overview of studies on experimental radioimmunotherapy. Cancer Res. 1995, 55, 5832s–5836s. [Google Scholar] [PubMed]
- Cheal, S.M.; Xu, H.; Guo, H.-f.; Zanzonico, P.B.; Larson, S.M.; Cheung, N.-K. Preclinical evaluation of multistep targeting of diasialoganglioside GD2 using an IgG-scFv bispecific antibody with high affinity for GD2 and DOTA metal complex. Mol. Cancer Ther. 2014, 13, 1803–1812. [Google Scholar] [CrossRef]
- Sharkey, R.M.; Primus, F.J.; Goldenberg, D.M. Second antibody clearance of radiolabeled antibody in cancer radioimmunodetection. Proc. Natl. Acad. Sci. USA 1984, 81, 2843–2846. [Google Scholar] [CrossRef]
- Zeng, D.; Zeglis, B.M.; Lewis, J.S.; Anderson, C.J. The growing impact of bioorthogonal click chemistry on the development of radiopharmaceuticals. J. Nucl. Med. 2013, 54, 829–832. [Google Scholar] [CrossRef]
- Meyer, J.-P.; Adumeau, P.; Lewis, J.S.; Zeglis, B.M. Click Chemistry and Radiochemistry: The First 10 Years. Bioconjug. Chem. 2016, 27, 2791–2807. [Google Scholar] [CrossRef]
- Mohsin, H.; Jia, F.; Bryan, J.N.; Sivaguru, G.; Cutler, C.S.; Ketring, A.R.; Miller, W.H.; Simón, J.; Frank, R.K.; Theodore, L.J. Comparison of pretargeted and conventional CC49 radioimmunotherapy using 149Pm, 166Ho, and 177Lu. Bioconjug. Chem. 2011, 22, 2444–2452. [Google Scholar] [CrossRef]
- Greenwood, F.C.; Hunter, W.M.; Glover, J.S. The Preparation of I-131-Labelled Human Growth Hormone of High Specific Radioactivity. Biochem. J. 1963, 89, 114–123. [Google Scholar] [CrossRef]
- Liu, Y.; Peterson, D.A.; Kimura, H.; Schubert, D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J. Neurochem. 1997, 69, 581–593. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Bertozzi, C.R. A FRET-Based Fluorogenic Phosphine for Live-Cell Imaging with the Staudinger Ligation. Angew. Chem. 2008, 120, 2428–2431. [Google Scholar] [CrossRef]
- Chang, S.; Lamm, S.H. Human health effects of sodium azide exposure: A literature review and analysis. Int. J. Toxicol. 2003, 22, 175–186. [Google Scholar] [CrossRef]
- Staudinger, H.; Meyer, J. Ueber neue organische Phosphorverbindungen II. Phosphazine. Helv. Chim. Acta 2004, 2, 619–635. [Google Scholar] [CrossRef]
- Vugts, D.J.; Vervoort, A.; Stigter-van Walsum, M.; Visser, G.W.; Robillard, M.S.; Versteegen, R.M.; Vulders, R.C.; Herscheid, J.K.; van Dongen, G.A. Synthesis of phosphine and antibody-azide probes for in vivo Staudinger ligation in a pretargeted imaging and therapy approach. Bioconjug. Chem. 2011, 22, 2072–2081. [Google Scholar] [CrossRef]
- Lub-de Hooge, M.N.; Kosterink, J.G.; Perik, P.J.; Nijnuis, H.; Tran, L.; Bart, J.; Suurmeijer, A.J.; de Jong, S.; Jager, P.L.; de Vries, E.G. Preclinical characterisation of 111In-DTPA-trastuzumab. Br. J. Pharm. 2004, 143, 99–106. [Google Scholar] [CrossRef]
- Liu, G.; Dou, S.; Chen, X.; Chen, L.; Liu, X.; Rusckowski, M.; Hnatowich, D.J. Adding a clearing agent to pretargeting does not lower the tumor accumulation of the effector as predicted. Cancer Biother. Radiopharm. 2010, 25, 757–762. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soni, N.; Sarkar, S.; Bhise, A.; Ha, Y.S.; Park, W.; Yu, A.-R.; Kumar, V.; Lim, J.E.; Yoon, Y.-R.; Yoo, J. “Click-to-Clear”: A Strategy to Minimize Radioactivity from the Blood Pool Utilizing Staudinger Ligation. Pharmaceutics 2023, 15, 719. https://doi.org/10.3390/pharmaceutics15030719
Soni N, Sarkar S, Bhise A, Ha YS, Park W, Yu A-R, Kumar V, Lim JE, Yoon Y-R, Yoo J. “Click-to-Clear”: A Strategy to Minimize Radioactivity from the Blood Pool Utilizing Staudinger Ligation. Pharmaceutics. 2023; 15(3):719. https://doi.org/10.3390/pharmaceutics15030719
Chicago/Turabian StyleSoni, Nisarg, Swarbhanu Sarkar, Abhinav Bhise, Yeong Su Ha, Wonchoul Park, A-Ram Yu, Virendra Kumar, Jeong Eun Lim, Young-Ran Yoon, and Jeongsoo Yoo. 2023. "“Click-to-Clear”: A Strategy to Minimize Radioactivity from the Blood Pool Utilizing Staudinger Ligation" Pharmaceutics 15, no. 3: 719. https://doi.org/10.3390/pharmaceutics15030719
APA StyleSoni, N., Sarkar, S., Bhise, A., Ha, Y. S., Park, W., Yu, A.-R., Kumar, V., Lim, J. E., Yoon, Y.-R., & Yoo, J. (2023). “Click-to-Clear”: A Strategy to Minimize Radioactivity from the Blood Pool Utilizing Staudinger Ligation. Pharmaceutics, 15(3), 719. https://doi.org/10.3390/pharmaceutics15030719