

Block Copolymer Micelles Encapsulating Au(III) Bis(Dithiolene) Complexes as Promising Nanostructures with Antiplasmodial Activity


 , , and
, , and 
Abstract
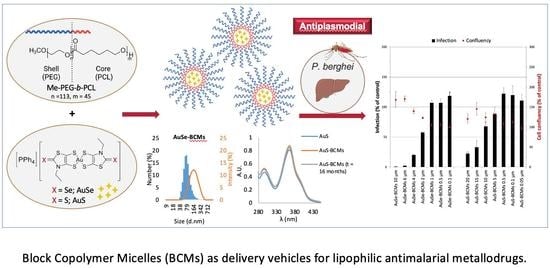
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.3. Synthesis and Characterization of MePEG-b-PCL
2.4. Synthesis of Micelles
2.5. Characterization of Micelles
2.6. Drug Loading Content and Efficiency
2.7. Stability Studies
2.8. In Vitro Release Studies
2.9. In Vitro Activity of Au(III) Bis(Dithiolene) Complexes on the Hepatic Stage of P. berghei Infection
3. Results and Discussion
3.1. Synthesis and Characterization of Micelles
3.2. Drug Loading Content and Efficiency
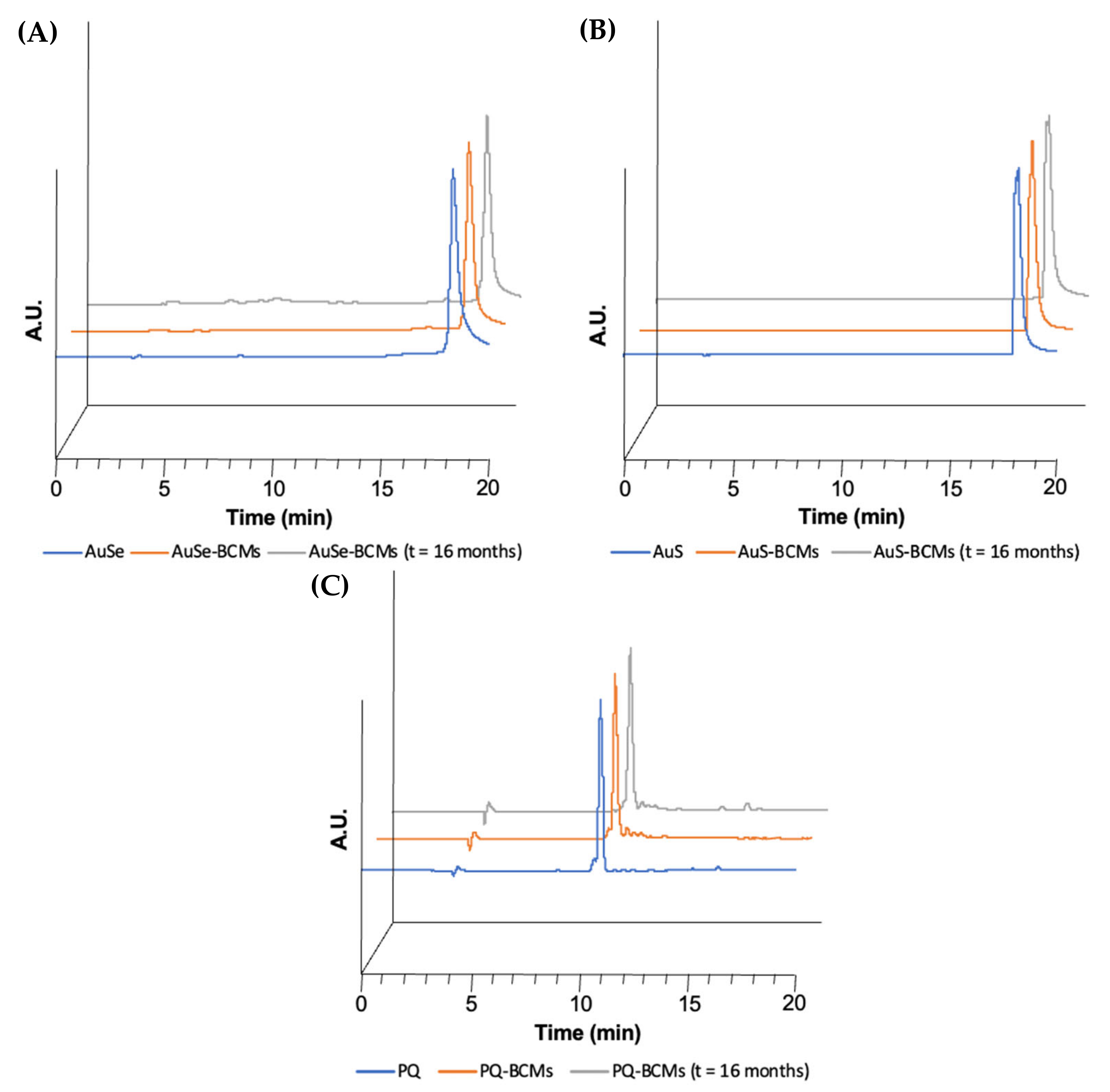
3.3. Stability Studies
3.4. In Vitro Release Studies
3.5. In Vitro Activity of Au(III) Bis(Dithiolene) Complexes on the Hepatic Stage of P. berghei Infection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- WHO. World Malaria Report; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Birkholtz, L.-M.; Bornman, R.; Focke, W.; Mutero, C.; de Jager, C. Sustainable malaria control: Transdisciplinary approaches for translational applications. Malar. J. 2012, 11, 431. [Google Scholar] [CrossRef]
- Ridley, R.G. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature 2002, 415, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Recht, J.; White, N.J. Primaquine: The risks and the benefits. Malar. J. 2014, 13, 418. [Google Scholar] [CrossRef]
- Asua, V.; Conrad, M.D.; Aydemir, O.; Duvalsaint, M.; Legac, J.; Duarte, E.; Tumwebaze, P.; Chin, D.M.; Cooper, R.A.; Yeka, A.; et al. Changing Prevalence of Potential Mediators of Aminoquinoline, Antifolate, and Artemisinin Resistance Across Uganda. J. Infect. Dis. 2020, 223, 985–994. [Google Scholar] [CrossRef]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.-L.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.-B.; Munguti, K.; et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef]
- Yasri, S.; Wiwanitkit, V. Artemisinin resistance: An important emerging clinical problem in tropical medicine. Int. J. Physiol. Pathophysiol. Pharmacol. 2021, 13, 152–157. [Google Scholar]
- Sannella, A.R.; Casini, A.; Gabbiani, C.; Messori, L.; Bilia, A.R.; Vincieri, F.F.; Majori, G.; Severini, C. New uses for old drugs. Auranofin, a clinically established antiarthritic metallodrug, exhibits potent antimalarial effects in vitro: Mechanistic and pharmacological implications. FEBS Lett. 2008, 582, 844–847. [Google Scholar] [CrossRef]
- Chaffman, M.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Auranofin: A Preliminary Review of its Pharmacological Properties and Therapeutic Use in Rheumatoid Arthritis. Drugs 1984, 27, 378–424. [Google Scholar] [CrossRef] [PubMed]
- Molter, A.; Rust, J.; Lehmann, C.W.; Deepa, G.; Chiba, P.; Mohr, F. Synthesis, structures and anti-malaria activity of some gold(i) phosphine complexes containing seleno- and thiosemicarbazonato ligands. Dalton Trans. 2011, 40, 9810–9820. [Google Scholar] [CrossRef] [PubMed]
- Khanye, S.D.; Wan, B.; Franzblau, S.G.; Gut, J.; Rosenthal, P.J.; Smith, G.S.; Chibale, K. Synthesis and in vitro antimalarial and antitubercular activity of gold(III) complexes containing thiosemicarbazone ligands. J. Organomet. Chem. 2011, 696, 3392–3396. [Google Scholar] [CrossRef]
- Ssemaganda, A.; Low, L.M.; Verhoeft, K.R.; Wambuzi, M.; Kawoozo, B.; Nabasumba, S.B.; Mpendo, J.; Bagaya, B.S.; Kiwanuka, N.; Stanisic, D.I.; et al. Gold(i) phosphine compounds as parasite attenuating agents for malaria vaccine and drug development. Metallomics 2018, 10, 444–454. [Google Scholar] [CrossRef]
- Hemmert, C.; Fabié, A.; Fabre, A.; Benoit-Vical, F.; Gornitzka, H. Synthesis, structures, and antimalarial activities of some silver(I), gold(I) and gold(III) complexes involving N-heterocyclic carbene ligands. Eur. J. Med. Chem. 2013, 60, 64–75. [Google Scholar] [CrossRef]
- Le Gal, Y.; Filatre-Furcate, A.; Lorcy, D.; Jeannin, O.; Roisnel, T.; Dorcet, V.; Fontinha, D.; Francisco, D.; Prudncio, M.; Martins, M.; et al. Broad Spectrum Functional Activity of Structurally Related Monoanionic Au(III) Bis(Dithiolene) Complexes. Int. J. Mol. Sci. 2022, 23, 7146. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surfaces B Biointerfaces 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Manjili, H.R.K.; Malvandi, H.; Mosavi, M.-S.; Danafar, H.; Reza, K.M.H.; Hojjat, M.; Mir-Sajjad, M.; Hossein, D. Preparation and Physicochemical Characterization of Biodegradable mPEG-PCL Core-Shell Micelles for Delivery of Artemisinin. Pharm. Sci. 2016, 22, 234–243. [Google Scholar] [CrossRef]
- Ramazani, A.; Keramati, M.; Malvandi, H.; Danafar, H.; Manjili, H.K. Preparation and in vivo evaluation of anti-plasmodial properties of artemisinin-loaded PCL–PEG–PCL nanoparticles. Pharm. Dev. Technol. 2017, 23, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Rashidzadeh, H.; Zamani, P.; Amiri, M.; Hassanzadeh, S.M.; Ramazani, A. Nanoincorporation of Plumbagin in Micelles Increase Its In Vivo Anti-Plasmodial Properties. Iran. J. Parasitol. 2022, 17, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Hyun, H.; Cho, Y.H.; Seo, K.S.; Jang, W.Y.; Kim, S.K.; Khang, G.; Lee, H.B. Preparation of methoxy poly(ethyleneglycol)-block-poly(caprolactone) via activated monomer mechanism and examination of micellar characterization. Polym. Bull. 2005, 55, 149–156. [Google Scholar] [CrossRef]
- Lou, X.; Detrembleur, C.; Jérôme, R. Living Cationic Polymerization of δ-Valerolactone and Synthesis of High Molecular Weight Homopolymer and Asymmetric Telechelic and Block Copolymer. Macromolecules 2002, 35, 1190–1195. [Google Scholar] [CrossRef]
- Zeng, F.; Lee, H.; Allen, C. Epidermal Growth Factor-Conjugated Poly(ethylene glycol)-block- Poly(δ-valerolactone) Copolymer Micelles for Targeted Delivery of Chemotherapeutics. Bioconjug. Chem. 2006, 17, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.; Lee, H.; Reilly, R.M.; Allen, C. Noninvasive Monitoring of the Fate of 111In-Labeled Block Copolymer Micelles by High Resolution and High Sensitivity MicroSPECT/CT Imaging. Mol. Pharm. 2009, 6, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.; Reilly, R.M.; Allen, C. Block Copolymer Micelles Target Auger Electron Radiotherapy to the Nucleus of HER2-Positive Breast Cancer Cells. Biomacromolecules 2012, 13, 455–465. [Google Scholar] [CrossRef]
- Zhao, J.; Wu, C.; Abbruzzese, J.; Hwang, R.F.; Li, C. Cyclopamine-Loaded Core-Cross-Linked Polymeric Micelles Enhance Radiation Response in Pancreatic Cancer and Pancreatic Stellate Cells. Mol. Pharm. 2015, 12, 2093–2100. [Google Scholar] [CrossRef]
- Lincha, V.R.; Zhao, J.; Wen, X.; Xiong, C.; Chow, D.S.-L.; Li, C. A polymeric micellar drug delivery system developed through a design of Experiment approach improves pancreatic tumor accumulation of calcipotriol and paclitaxel. Int. J. Pharm. 2021, 601, 120523. [Google Scholar] [CrossRef]
- Ploemen, I.H.J.; Prudêncio, M.; Douradinha, B.; Ramesar, J.; Fonager, J.; Van Gemert, G.-J.; Luty, A.; Hermsen, C.C.; Sauerwein, R.W.; Baptista, F.G.; et al. Visualisation and Quantitative Analysis of the Rodent Malaria Liver Stage by Real Time Imaging. PLoS ONE 2009, 4, e7881. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.; Alho, I.; Marques, F.; Gano, L.; Correia, I.; Correia, J.D.; Casimiro, S.; Costa, L.; Santos, I.; Fernandes, C. Radiolabeled block copolymer micelles for image-guided drug delivery. Int. J. Pharm. 2016, 515, 692–701. [Google Scholar] [CrossRef]
- Yokoyama, M. Clinical Applications of Polymeric Micelle Carrier Systems in Chemotherapy and Image Diagnosis of Solid Tumors. J. Exp. Clin. Med. 2011, 3, 151–158. [Google Scholar] [CrossRef]
- Ribeiro, E.; Marques, F.; Gano, L.; Correia, J.D.; Santos, I.; Fernandes, C. Docetaxel-loaded block copolymer micelles labeled with 188Re for combined radiochemotherapy. J. Drug Deliv. Sci. Technol. 2020, 60, 101898. [Google Scholar] [CrossRef]
- Nosrati, H.; Barzegari, P.; Danafar, H.; Manjili, H.K. Biotin-functionalized copolymeric PEG-PCL micelles for in vivo tumour-targeted delivery of artemisinin. Artif. Cells Nanomed. Biotechnol. 2019, 47, 104–114. [Google Scholar] [CrossRef]
- Palma, G.; Conte, C.; Barbieri, A.; Bimonte, S.; Luciano, A.; Rea, D.; Ungaro, F.; Tirino, P.; Quaglia, F.; Arra, C. Antitumor activity of PEGylated biodegradable nanoparticles for sustained release of docetaxel in triple-negative breast cancer. Int. J. Pharm. 2014, 473, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lavasanifar, A.; Samuel, J.; Kwon, G.S. Poly(ethylene oxide)-block-poly(l-amino acid) micelles for drug delivery. Adv. Drug Deliv. Rev. 2002, 54, 169–190. [Google Scholar] [CrossRef]
- Yu, M.; Yuan, W.; Li, D.; Schwendeman, A.; Schwendeman, S.P. Predicting drug release kinetics from nanocarriers inside dialysis bags. J. Control. Release 2019, 315, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Tsong, Y.; Hammerstrom, T.; Chen, J.J. Multipoint dissolution specification and acceptance sampling rule based on profile modeling and principal component analysis. J. Biopharm. Stat. 1997, 7, 423–439. [Google Scholar] [CrossRef]
- Vale, N.; Moreira, R.; Gomes, P. Primaquine revisited six decades after its discovery. Eur. J. Med. Chem. 2009, 44, 937–953. [Google Scholar] [CrossRef]
- Prudêncio, M.; Mota, M.M.; Mendes, A.M. A toolbox to study liver stage malaria. Trends Parasitol. 2011, 27, 565–574. [Google Scholar] [CrossRef]
- Ramos-Inza, S.; Plano, D.; Sanmartín, C. Metal-based compounds containing selenium: An appealing approach towards novel therapeutic drugs with anticancer and antimicrobial effects. Eur. J. Med. Chem. 2022, 244, 114834. [Google Scholar] [CrossRef]
- Zhang, Y.; Sauer, A.; Parnham, M.J. Antimalarial properties of ebselen. Parasitol. Res. 1989, 75, 353–360. [Google Scholar] [CrossRef]
- Rashidi, S.; Fernández-Rubio, C.; Mansouri, R.; Ali-Hassanzadeh, M.; Ghani, E.; Karimazar, M.; Manzano-Román, R.; Nguewa, P. Selenium and protozoan parasitic infections: Selenocompounds and selenoproteins potential. Parasitol. Res. 2022, 121, 49–62. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Micelles | Dh (nm) | PDI | Zeta Potential (mV) | LC (%) | LE (%) |
|---|---|---|---|---|---|
| BCMs | 82.1 ± 2.7 | 0.21 ± 0.02 | −41.1 ± 0.2 | - | - |
| PQ-BCMs | 50.9 ± 2.8 | 0.22 ± 0.01 | −43.6 ± 1.6 | 3.7 ± 0.3 | 82.5 ± 0.7 |
| AuSe-BCMs | 87.1 ± 9.7 | 0.18 ± 0.004 | −38.6 ± 0.8 | 2.9 ± 0.3 | 55.5 ± 0.7 |
| AuS-BCMs | 72.8 ± 3.1 | 0.18 ± 0.02 | −57.3 ± 0.7 | 3.8 ± 0.4 | 77.4 ± 0.8 |
| Micelles | LC (%) t = 0 | LC (%) t = 16 Months |
|---|---|---|
| PQ-BCMs | 3.7 ± 0.3 | 3.8 ± 0.6 |
| AuSe-BCMs | 2.9 ± 0.3 | 2.8 ± 0.02 |
| AuS-BCMs | 3.8 ± 0.4 | 3.7 ± 0.1 |
| Parameters | PQ-BCMs | AuSe-BCMs | AuS-BCMs |
|---|---|---|---|
| k | 0.61 | 0.05 | 0.11 |
| Fmax | 91.49 | 86.66 | 58.12 |
| R2 | 0.98 | 0.89 | 0.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.F.; Azevedo, R.; Prudêncio, M.; Marques, F.; Le Gal, Y.; Lorcy, D.; Fernandes, C. Block Copolymer Micelles Encapsulating Au(III) Bis(Dithiolene) Complexes as Promising Nanostructures with Antiplasmodial Activity. Pharmaceutics 2023, 15, 1030. https://doi.org/10.3390/pharmaceutics15031030
Santos JF, Azevedo R, Prudêncio M, Marques F, Le Gal Y, Lorcy D, Fernandes C. Block Copolymer Micelles Encapsulating Au(III) Bis(Dithiolene) Complexes as Promising Nanostructures with Antiplasmodial Activity. Pharmaceutics. 2023; 15(3):1030. https://doi.org/10.3390/pharmaceutics15031030
Chicago/Turabian StyleSantos, Joana F., Raquel Azevedo, Miguel Prudêncio, Fernanda Marques, Yann Le Gal, Dominique Lorcy, and Célia Fernandes. 2023. "Block Copolymer Micelles Encapsulating Au(III) Bis(Dithiolene) Complexes as Promising Nanostructures with Antiplasmodial Activity" Pharmaceutics 15, no. 3: 1030. https://doi.org/10.3390/pharmaceutics15031030
APA StyleSantos, J. F., Azevedo, R., Prudêncio, M., Marques, F., Le Gal, Y., Lorcy, D., & Fernandes, C. (2023). Block Copolymer Micelles Encapsulating Au(III) Bis(Dithiolene) Complexes as Promising Nanostructures with Antiplasmodial Activity. Pharmaceutics, 15(3), 1030. https://doi.org/10.3390/pharmaceutics15031030