
Development of 3D Printed Multi-Layered Orodispersible Films with Porous Structure Applicable as a Substrate for Inkjet Printing

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
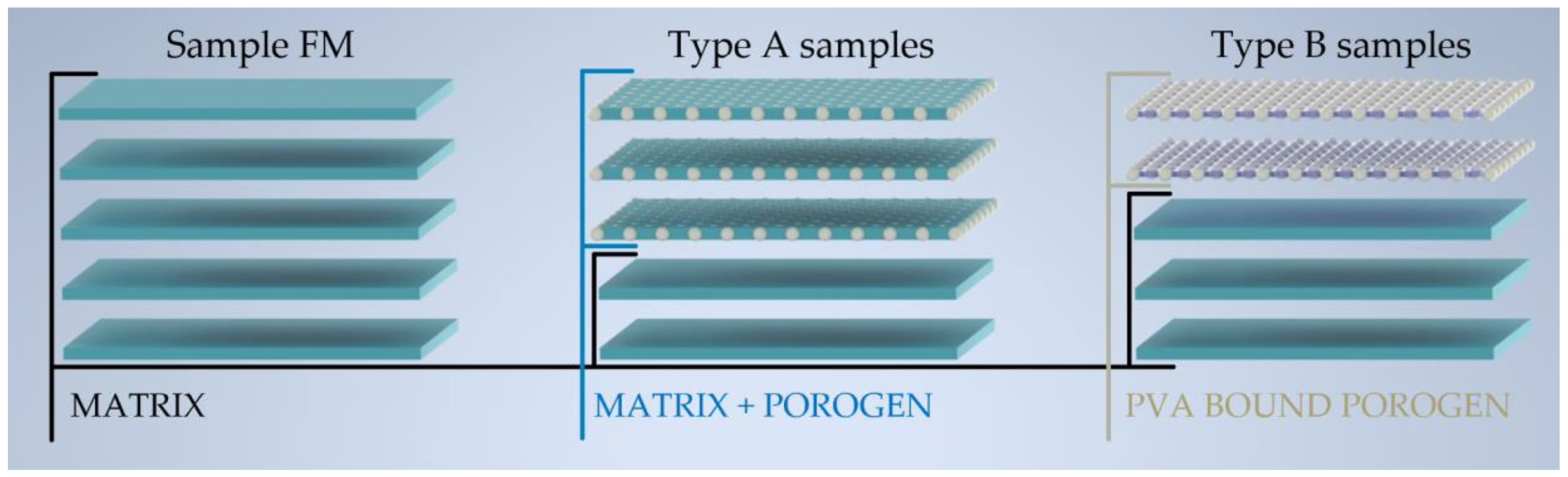
2.1. Materials
2.2. Print Dispersion Preparation and Viscosity Evaluation
2.3. SSE 3D Printing
2.4. Weight
2.5. Thickness
2.6. Mechanical Properties
2.7. Disintegration Time
2.8. Micro-CT
2.9. Scanning Electron Microscopy
3. Results and Discussions
3.1. SSE 3D Printing, Viscosity Evaluation
3.2. Weight
3.3. Thickness
3.4. Mechanical Properties
3.5. Disintegration Time
3.6. Micro-CT
3.7. Scanning Electron Microscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Planchette, C.; Pichler, H.; Wimmer-Teubenbacher, M.; Gruber, M.; Gruber-Woelfler, H.; Mohr, S.; Tetyczka, C.; Hsiao, W.-K.; Paudel, A.; Roblegg, E.; et al. Printing Medicines as Orodispersible Dosage Forms: Effect of Substrate on the Printed Micro-Structure. Int. J. Pharm. 2016, 509, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Knowles, L.; Luth, W.; Bubela, T. Paving the road to personalized medicine: Recommendations on regulatory, intellectual property and reimbursement challenges. J. Law Biosci. 2017, 4, 453–506. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.S. Ways to Minimize Adverse Drug Reactions. Postgrad. Med. 1999, 106, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, D.E.; Moeckel, F.; Villa, M.S.; Housman, L.T.; McCarty, C.A.; McLeod, H.L. Strategies for integrating personalized medicine into healthcare practice. Pers. Med. 2017, 14, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Jamróz, W.; Kurek, M.; Łyszczarz, E.; Szafraniec, J.; Knapik-Kowalczuk, J.; Syrek, K.; Paluch, M.; Jachowicz, R. 3D Printed Orodispersible Films with Aripiprazole. Int. J. Pharm. 2017, 533, 413–420. [Google Scholar] [CrossRef]
- Sjöholm, E.; Sandler, N. Additive Manufacturing of Personalized Orodispersible Warfarin Films. Int. J. Pharm. 2019, 564, 117–123. [Google Scholar] [CrossRef]
- Roden, D.F.; Altman, K.W. Causes of Dysphagia Among Different Age Groups: A Systematic Review of the Literature. Otolaryngol. Clin. N. Am. 2013, 46, 965–987. [Google Scholar] [CrossRef]
- Breitkreutz, J.; Boos, J. Paediatric and Geriatric Drug Delivery. Expert Opin. Drug Deliv. 2007, 4, 37–45. [Google Scholar] [CrossRef]
- Preis, M.; Pein, M.; Breitkreutz, J. Development of a Taste-Masked Orodispersible Film Containing Dimenhydrinate. Pharmaceutics 2012, 4, 551–562. [Google Scholar] [CrossRef]
- Panraksa, P.; Tipduangta, P.; Jantanasakulwong, K.; Jantrawut, P. Formulation of Orally Disintegrating Films as an Amorphous Solid Solution of a Poorly Water-Soluble Drug. Membranes 2020, 10, 376. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nishimatsu, T.; Tahara, K.; Takeuchi, H. Novel Use of Insoluble Particles as Disintegration Enhancers for Orally Disintegrating Films. J. Drug Deliv. Sci. Technol. 2019, 54, 101310. [Google Scholar] [CrossRef]
- Cilurzo, F.; Musazzi, U.M.; Franzé, S.; Selmin, F.; Minghetti, P. Orodispersible dosage forms: Biopharmaceutical improvements and regulatory requirements. Drug Discov. Today 2018, 23, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Slavkova, M.; Breitkreutz, J. Orodispersible drug formulations for children and elderly. Eur. J. Pharm. Sci. 2015, 75, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Genina, N.; Janßen, E.M.; Breitenbach, A.; Breitkreutz, J.; Sandler, N. Evaluation of Different Substrates for Inkjet Printing of Rasagiline Mesylate. Eur. J. Pharm. Biopharm. 2013, 85, 1075–1083. [Google Scholar] [CrossRef]
- Hoffmann, E.M.; Breitenbach, A.; Breitkreutz, J. Advances in Orodispersible Films for Drug Delivery. Expert Opin. Drug Deliv. 2011, 8, 299–316. [Google Scholar] [CrossRef]
- Dixit, R.P.; Puthli, S.P. Oral Strip Technology: Overview and Future Potential. J. Control. Release 2009, 139, 94–107. [Google Scholar] [CrossRef]
- Janßen, E.M.; Schliephacke, R.; Breitenbach, A.; Breitkreutz, J. Drug-Printing by Flexographic Printing Technology—A New Manufacturing Process for Orodispersible Films. Int. J. Pharm. 2013, 441, 818–825. [Google Scholar] [CrossRef]
- Seoane-Viaño, I.; Januskaite, P.; Alvarez-Lorenzo, C.; Basit, A.W.; Goyanes, A. Semi-solid extrusion 3D printing in drug delivery and biomedicine: Personalised solutions for healthcare challenges. J Control. Release 2021, 332, 367–389. [Google Scholar] [CrossRef]
- Iftimi, L.-D.; Edinger, M.; Bar-Shalom, D.; Rantanen, J.; Genina, N. Edible Solid Foams as Porous Substrates for Inkjet-Printable Pharmaceuticals. Eur. J. Pharm. Biopharm. 2019, 136, 38–47. [Google Scholar] [CrossRef]
- Edinger, M.; Bar-Shalom, D.; Sandler, N.; Rantanen, J.; Genina, N. QR Encoded Smart Oral Dosage Forms by Inkjet Printing. Int. J. Pharm. 2018, 536, 138–145. [Google Scholar] [CrossRef]
- Borges, A.F.; Silva, C.; Coelho, J.F.J.; Simões, S. Oral Films: Current Status and Future Perspectives: I—Galenical Development and Quality Attributes. J. Control. Release 2015, 206, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Alomari, M.; Mohamed, F.H.; Basit, A.W.; Gaisford, S. Personalised Dosing: Printing a Dose of One’s Own Medicine. Int. J. Pharm. 2015, 494, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Raijada, D.; Genina, N.; Fors, D.; Wisaeus, E.; Peltonen, J.; Rantanen, J.; Sandler, N. A Step Toward Development of Printable Dosage Forms for Poorly Soluble Drugs. J. Pharm. Sci. 2013, 102, 3694–3704. [Google Scholar] [CrossRef] [PubMed]
- Vraníková, B.; Niederquell, A.; Šklubalová, Z.; Kuentz, M. Relevance of the Theoretical Critical Pore Radius in Mesoporous Silica for Fast Crystallizing Drugs. Int. J. Pharm. 2020, 591, 120019. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Yamakawa, R.; Nishimatsu, T.; Takeuchi, Y.; Hayakawa, K.; Maruyama, N. Design of Rapidly Disintegrating Drug Delivery Films for Oral Doses with Hydoxypropyl Methylcellulose. J. Drug Deliv. Sci. Technol. 2013, 23, 471–475. [Google Scholar] [CrossRef]
- Elbl, J.; Gajdziok, J.; Kolarczyk, J. 3D Printing of Multilayered Orodispersible Films with In-Process Drying. Int. J. Pharm. 2020, 575, 118883. [Google Scholar] [CrossRef]
- Janigová, N.; Elbl, J.; Pavloková, S.; Gajdziok, J. Effects of Various Drying Times on the Properties of 3D Printed Orodispersible Films. Pharmaceutics 2022, 14, 250. [Google Scholar] [CrossRef]
- Preis, M.; Gronkowsky, D.; Grytzan, D.; Breitkreutz, J. Comparative study on novel test systems to determine disintegration time of orodispersible films. J. Pharm. Pharmacol. 2014, 66, 1102–1111. [Google Scholar] [CrossRef]
- Preis, M.; Knop, K.; Breitkreutz, J. Mechanical Strength Test for Orodispersible and Buccal Films. Int. J. Pharm. 2014, 461, 22–29. [Google Scholar] [CrossRef]
- Gupta, M.S.; Kumar, T.P.; Gowda, D.V.; Rosenholm, J.M. Orodispersible Films: Conception to Quality by Design. Adv. Drug Deliv. Rev. 2021, 178, 113983. [Google Scholar] [CrossRef]
- Rycerz, K.; Stepien, K.A.; Czapiewska, M.; Arafat, B.T.; Habashy, R.; Isreb, A.; Peak, M.; Alhnan, M.A. Embedded 3D Printing of Novel Bespoke Soft Dosage Form Concept for Pediatrics. Pharmaceutics 2019, 11, 630. [Google Scholar] [CrossRef] [PubMed]
- FDA. Orally Disintegrating Tablets. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/orally-disintegrating-tablets (accessed on 12 December 2022).
- European Pharmacopoeia 10.8. Available online: https://www.edqm.eu/en/-/european-pharmacopoeia-supplement-10.8-now-available (accessed on 12 December 2022).
- Liew, K.B.; Tan, Y.T.F.; Peh, K.K. Effect of polymer, plasticizer and filler on orally disintegrating film. Drug Dev. Ind. Pharm. 2014, 40, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Buanz, A.B.M.; Saunders, M.H.; Basit, A.W.; Gaisford, S. Preparation of Personalized-Dose Salbutamol Sulphate Oral Films with Thermal Ink-Jet Printing. Pharm. Res. 2011, 28, 2386–2392. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition Type | Sample | Excipient Concentration in Dispersion (wt%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PVA | HPMC | Gly | Ex + Et | W | X | Porogen | No of Layers | ||
| Bottom layer matrix | FM | 5 | 1.5 | 5 | 2.5 + 2.5 | 83.5 | - | X | 2 or 3 |
| Type A top layer | *0.5 | 5 | 1.5 | 5 | 2.5 + 2.5 | 83.0 | - | 0.5 | 3 |
| *1 | 5 | 1.5 | 5 | 2.5 + 2.5 | 82.5 | - | 1.0 | 3 | |
| *1.5 | 5 | 1.5 | 5 | 2.5 + 2.5 | 82.0 | - | 1.5 | 3 | |
| Type B top layer | X*2.5 | 1 | - | - | - | 96.0625 | 0.4375 | 2.5 | 2 |
| X*5 | 1 | - | - | - | 93.5750 | 0.4250 | 5.0 | 2 | |
| Composition Type | Sample | Viscosity (cP) | AVG Weight (mg) | RSD (%) | AVG Thickness (µm) | RSD (%) |
|---|---|---|---|---|---|---|
| MATRIX | FM | 57.10 | 88.0 ± 3.80 | 4.32 | 124.11 ± 17.75 | 14.26 |
| A | A0.5 | 67.25 | 83.8 ± 0.64 | 0.76 | 130.86 ± 30.70 | 23.46 |
| A1 | 96.50 | 86.1 ± 0.51 | 0.59 | 134.62 ± 30.50 | 22.66 | |
| A1.5 | 118.00 | 85.7 ± 1.27 | 1.48 | 125.24 ± 27.00 | 21.56 | |
| F0.5 | 98.25 | 80.6 ± 0.58 | 0.72 | 107.25 ± 17.00 | 15.85 | |
| F1 | 108.50 | 80.9 ± 0.67 | 0.83 | 114.15 ± 17.19 | 15.06 | |
| F1.5 | 116.80 | 83.9 ± 0.73 | 0.87 | 128.93 ± 22.78 | 17.67 | |
| NS0.5 | 79.25 | 80.5 ± 0.66 | 0.82 | 110.04 ± 19.86 | 18.05 | |
| NS1 | 112.80 | 83.8 ± 0.68 | 0.81 | 120.61 ± 19.67 | 16.31 | |
| NS1.5 | 195.80 | 83.9 ± 0.73 | 0.87 | 115.78 ± 17.38 | 15.01 | |
| NUS0.5 | 122.00 | 78.8 ± 1.73 | 2.19 | 107.87 ± 17.93 | 16.62 | |
| NUS1 | 127.30 | 79.8 ± 0.73 | 0.92 | 131.77 ± 21.47 | 16.29 | |
| NUS1.5 | 136.50 | 83.2 ± 2.62 | 3.15 | 149.87 ± 33.18 | 22.14 | |
| NUFL0.5 | 109.50 | 86.4 ± 2.03 | 2.35 | 115.16 ± 19.21 | 16.68 | |
| NUFL1 | 174.30 | 85.2 ± 2.01 | 2.36 | 115.26 ± 17.61 | 15.28 | |
| NUFL1.5 | 216.80 | 91.1 ± 3.17 | 3.48 | 136.78 ± 23.21 | 16.97 | |
| SFP0.5 | 76.75 | 86.3 ± 0.81 | 0.94 | 111.12 ± 14.06 | 12.65 | |
| SFP1 | 124.30 | 90.0 ± 0.98 | 1.09 | 112.21 ± 21.30 | 18.98 | |
| SFP1.5 | 187.50 | 90.4 ± 0.89 | 0.99 | 110.78 ± 18.08 | 16.32 | |
| SX0.5 | 69.25 | 86.6 ± 1.10 | 1.27 | 102.92 ± 15.67 | 15.23 | |
| SX1 | 89.25 | 86.9 ± 1.46 | 1.68 | 105.14 ± 10.73 | 10.21 | |
| SX1.5 | 111.00 | 89.7 ± 1.46 | 1.63 | 127.05 ± 20.66 | 16.26 | |
| B | XA2.5 | 71.00 | 61.6 ± 0.68 | 1.11 | 77.34 ± 7.86 | 10.16 |
| XA5 | 95.00 | 63.1 ± 1.53 | 2.43 | 96.73 ± 10.98 | 11.35 | |
| XF2.5 | 74.00 | 61.0 ± 2.95 | 4.83 | 82.91 ± 10.53 | 12.70 | |
| XF5 | 105.30 | 66.5 ± 1.96 | 2.94 | 113.59 ± 13.97 | 12.30 | |
| XNS2.5 | 76.75 | 62.4 ± 0.80 | 1.28 | 77.07 ± 10.38 | 13.47 | |
| XNS5 | 84.75 | 66.3 ± 1.33 | 2.01 | 87.43 ± 11.86 | 13.57 | |
| XNUS2.5 | 73.25 | 56.3 ± 0.83 | 1.47 | 79.92 ± 10.31 | 12.90 | |
| XNUFL2.5 | 77.00 | 59.7 ± 0.46 | 0.77 | 78.26 ± 10.92 | 13.95 | |
| XSFP2.5 | 88.50 | 62.4 ± 0.34 | 0.54 | 79.36 ± 15.13 | 19.06 | |
| XSX2.5 | 67.25 | 61.9 ± 0.58 | 0.93 | 78.50 ± 8.81 | 11.22 | |
| XSX5 | 94.25 | 67.6 ± 0.55 | 0.81 | 114.47 ± 16.17 | 14.13 |
| Composition Type | Sample | Tensile Strength (N/mm2) | SD (N/mm2) | Elongation at Break (%) | SD (%) |
|---|---|---|---|---|---|
| MATRIX | FM | 7.07 | 0.24 | 45.80 | 10.56 |
| A | A0.5 | 2.99 | 0.21 | 59.00 | 8.23 |
| A1 | 3.09 | 0.11 | 55.50 | 4.83 | |
| A1.5 | 3.70 | 0.11 | 59.60 | 6.69 | |
| F0.5 | 2.64 | 0.11 | 30.80 | 3.12 | |
| F1 | 2.43 | 0.18 | 23.00 | 2.00 | |
| F1.5 | 2.11 | 0.14 | 19.30 | 3.20 | |
| NS0.5 | 3.50 | 0.07 | 50.70 | 5.10 | |
| NS1 | 3.39 | 0.08 | 46.40 | 2.07 | |
| NS1.5 | 3.94 | 0.13 | 46.40 | 6.84 | |
| NUS0.5 | 3.97 | 0.12 | 52.20 | 6.36 | |
| NUS1 | 3.25 | 0.14 | 37.80 | 3.21 | |
| NUS1.5 | 2.95 | 0.14 | 35.10 | 4.59 | |
| NUFL0.5 | 1.86 | 0.15 | 21.60 | 3.51 | |
| NUFL1 | 2.00 | 0.16 | 21.00 | 3.18 | |
| NUFL1.5 | 1.68 | 0.11 | 18.80 | 1.28 | |
| SFP0.5 | 2.12 | 0.19 | 24.90 | 3.10 | |
| SFP1 | 2.01 | 0.14 | 22.40 | 2.48 | |
| SFP1.5 | 2.07 | 0.04 | 24.90 | 2.16 | |
| SX0.5 | 1.65 | 0.02 | 29.30 | 0.90 | |
| SX1 | 2.16 | 0.06 | 29.50 | 0.83 | |
| SX1.5 | 1.79 | 0.01 | 24.70 | 1.46 | |
| B | XA2.5 | 2.33 | 0.09 | 17.10 | 1.87 |
| XA5 | 1.13 | 0.05 | 16.00 | 1.86 | |
| XF2.5 | 1.56 | 0.10 | 11.70 | 1.32 | |
| XF5 | 0.85 | 0.28 | 7.70 | 2.57 | |
| XNS2.5 | 1.98 | 0.27 | 17.10 | 2.68 | |
| XNS5 | 1.63 | 0.06 | 11.10 | 0.79 | |
| XNUS2.5 | 1.27 | 0.07 | 12.50 | 0.28 | |
| XNUFL2.5 | 1.76 | 0.27 | 12.80 | 2.73 | |
| XSFP2.5 | 1.19 | 0.25 | 7.80 | 1.45 | |
| XSX2.5 | 1.99 | 0.30 | 13.30 | 3.13 | |
| XSX5 | 1.22 | 0.10 | 12.40 | 0.84 |
| Composition Type | Sample | Puncture Strength (N/mm2) | SD (N/mm2) | Elongation to Puncture (%) | SD (%) |
|---|---|---|---|---|---|
| MATRIX | FM | 1.95 | 0.26 | 48.51 | 6.56 |
| A | A0.5 | 1.09 | 0.07 | 21.34 | 1.16 |
| A1 | 0.89 | 0.06 | 16.51 | 0.55 | |
| A1.5 | 1.05 | 0.08 | 17.16 | 0.81 | |
| F0.5 | 0.6 | 0.03 | 9.69 | 0.44 | |
| F1 | 0.52 | 0.06 | 8.05 | 0.44 | |
| F1.5 | 0.48 | 0.05 | 6.61 | 0.36 | |
| NS0.5 | 0.88 | 0.14 | 18.87 | 1.34 | |
| NS1 | 0.64 | 0.01 | 9.84 | 0.35 | |
| NS1.5 | 0.58 | 0.08 | 8.75 | 0.69 | |
| NUS0.5 | 0.92 | 0.10 | 12.84 | 1.05 | |
| NUS1 | 0.94 | 0.07 | 12.1 | 0.56 | |
| NUS1.5 | 0.79 | 0.14 | 9.61 | 0.81 | |
| NUFL0.5 | 0.33 | 0.04 | 6.23 | 0.21 | |
| NUFL1 | 0.32 | 0.03 | 5.58 | 0.41 | |
| NUFL1.5 | 0.33 | 0.03 | 5.46 | 0.16 | |
| SFP0.5 | 0.49 | 0.03 | 8.96 | 0.35 | |
| SFP1 | 0.39 | 0.03 | 6.99 | 0.23 | |
| SFP1.5 | 0.36 | 0.01 | 7.25 | 0.27 | |
| SX0.5 | 0.31 | 0.02 | 8.32 | 0.18 | |
| SX1 | 0.38 | 0.02 | 7.91 | 0.24 | |
| SX1.5 | 0.40 | 0.03 | 7.58 | 0.43 | |
| B | XA2.5 | 0.22 | 0.03 | 4.21 | 0.49 |
| XA5 | 0.13 | 0.01 | 3.91 | 0.45 | |
| XF2.5 | 0.15 | 0.03 | 3.21 | 0.44 | |
| XF5 | 0.16 | 0.03 | 3.62 | 0.33 | |
| XNS2.5 | 0.22 | 0.02 | 4.68 | 0.30 | |
| XNS5 | 0.18 | 0.01 | 3.30 | 0.24 | |
| XNUS2.5 | 0.15 | 0.01 | 3.72 | 0.23 | |
| XNUFL2.5 | 0.21 | 0.01 | 4.53 | 0.21 | |
| XSFP2.5 | 0.14 | 0.05 | 2.94 | 0.65 | |
| XSX2.5 | 0.25 | 0.02 | 4.63 | 0.28 | |
| XSX5 | 0.25 | 0.03 | 5.18 | 0.40 |
| Composition Type | Sample | AVG DT (s) | AVG DT to 100 µm (s) | RSD (%) | Porosity |
|---|---|---|---|---|---|
| MATRIX | FM | 17.90 ± 1.57 | 14.42 ± 1.26 | 8.75 | 8.7 |
| A | A0.5 | 13.67 ± 0.80 | 10.45 ± 0.61 | 5.83 | X |
| A1 | 12.95 ± 0.47 | 9.62 ± 0.35 | 3.60 | x | |
| A1.5 | 12.23 ± 1.25 | 9.76 ± 1.00 | 10.24 | 1.76 | |
| F0.5 | 9.95 ± 1.53 | 9.27 ± 1.42 | 15.35 | x | |
| F1 | 10.50 ± 1.20 | 9.20 ± 1.05 | 11.41 | x | |
| F1.5 | 10.74 ± 1.14 | 8.33 ± 0.88 | 10.61 | 8.94 | |
| NS0.5 | 11.98 ± 0.53 | 10.88 ± 0.48 | 4.39 | x | |
| NS1 | 13.01 ± 0.65 | 10.79 ± 0.54 | 5.02 | x | |
| NS1.5 | 12.08 ± 0.80 | 10.43 ± 0.69 | 6.60 | 1.29 | |
| NUS0.5 | 9.82 ± 1.83 | 9.10 ± 1.70 | 18.69 | x | |
| NUS1 | 11.10 ± 0.38 | 8.43 ± 0.29 | 3.46 | x | |
| NUS1.5 | 9.25 ± 3.16 | 6.17 ± 2.11 | 34.17 | 2.24 | |
| NUFL0.5 | 11.50 ± 0.14 | 9.99 ± 0.12 | 1.22 | x | |
| NUFL1 | 11.33 ± 1.32 | 9.83 ± 1.15 | 11.66 | x | |
| NUFL1.5 | 10.94 ± 1.53 | 8.00 ± 1.12 | 13.99 | 7.38 | |
| SFP0.5 | 12.19 ± 0.66 | 10.97 ± 0.60 | 5.43 | x | |
| SFP1 | 10.46 ± 1.09 | 9.33 ± 0.97 | 10.40 | x | |
| SFP1.5 | 9.39 ± 0.42 | 8.48 ± 0.38 | 4.48 | 2.25 | |
| SX0.5 | 9.76 ± 0.41 | 9.48 ± 0.40 | 4.25 | x | |
| SX1 | 10.81 ± 0.88 | 10.28 ± 0.84 | 8.16 | x | |
| SX1.5 | 10.37 ± 0.54 | 8.17 ± 0.43 | 5.22 | 3.18 | |
| B | XA2.5 | 5.57 ± 0.28 | 7.20 ± 0.36 | 4.96 | x |
| XA5 | 3.69 ± 0.09 | 3.82 ± 0.09 | 2.42 | 17.64 | |
| XF2.5 | 4.61 ± 0.46 | 5.56 ± 0.56 | 10.04 | x | |
| XF5 | 3.46 ± 0.49 | 3.05 ± 0.43 | 14.17 | 22.76 | |
| XNS2.5 | 4.71 ± 0.46 | 6.12 ± 0.59 | 9.69 | x | |
| XNS5 | 5.47 ± 0.58 | 6.26 ± 0.67 | 10.66 | 16.75 | |
| XNUS2.5 | 3.55 ± 0.12 | 4.44 ± 0.15 | 3.31 | 10.36 | |
| XNUFL2.5 | 5.31 ± 0.33 | 6.78 ± 0.42 | 6.13 | 6.11 | |
| XSFP2.5 | 3.85 ± 0.51 | 4.86 ± 0.64 | 13.21 | 21.34 | |
| XSX2.5 | 5.45 ± 0.48 | 6.95 ± 0.61 | 8.83 | x | |
| XSX5 | 4.27 ± 0.31 | 3.73 ± 0.27 | 7.23 | 14.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbl, J.; Veselý, M.; Blaháčková, D.; Ondruš, J.; Kulich, P.; Mašková, E.; Mašek, J.; Gajdziok, J. Development of 3D Printed Multi-Layered Orodispersible Films with Porous Structure Applicable as a Substrate for Inkjet Printing. Pharmaceutics 2023, 15, 714. https://doi.org/10.3390/pharmaceutics15020714
Elbl J, Veselý M, Blaháčková D, Ondruš J, Kulich P, Mašková E, Mašek J, Gajdziok J. Development of 3D Printed Multi-Layered Orodispersible Films with Porous Structure Applicable as a Substrate for Inkjet Printing. Pharmaceutics. 2023; 15(2):714. https://doi.org/10.3390/pharmaceutics15020714
Chicago/Turabian StyleElbl, Jan, Martin Veselý, Dagmar Blaháčková, Jaroslav Ondruš, Pavel Kulich, Eliška Mašková, Josef Mašek, and Jan Gajdziok. 2023. "Development of 3D Printed Multi-Layered Orodispersible Films with Porous Structure Applicable as a Substrate for Inkjet Printing" Pharmaceutics 15, no. 2: 714. https://doi.org/10.3390/pharmaceutics15020714
APA StyleElbl, J., Veselý, M., Blaháčková, D., Ondruš, J., Kulich, P., Mašková, E., Mašek, J., & Gajdziok, J. (2023). Development of 3D Printed Multi-Layered Orodispersible Films with Porous Structure Applicable as a Substrate for Inkjet Printing. Pharmaceutics, 15(2), 714. https://doi.org/10.3390/pharmaceutics15020714